
Abstract
Background:
Adverse events for continuous glucose monitors (CGMs) represent a significant issue for people with diabetes with 281 963 CGM adverse events occurring in 2022. The process to obtain adverse events and the US Food and Drug Administration (FDA) database that contains them are reviewed.
Methods:
Tables were created in SQL Server for four CGM products (Dexcom G6, all versions of Abbott Libre, Medtronic Guardian 3, and Senseonics Eversense) containing either malfunction or injury adverse events sorted by the manufacturer’s chosen product code. As the product code is not always clear (or appropriate), the causes of the events were determined from the text description of the adverse event. The resulting causes were listed in decreasing order in tables for each product and event type.
Results:
A common effect of several event causes prevented the user from obtaining a result. Inaccuracy was also a frequent complaint. Other causes were specific to that device.
Conclusions:
Creating tables based on manufacturer problem codes for their CGMs, followed by analysis of the adverse event text, facilitates the analysis of event causes. Analyzing adverse event data is the first step in trying to reduce the number of adverse events.
Introduction
Continuous glucose monitors (CGMs) are important in the treatment of diabetes. 1 Yet, adverse events for CGMs represent a significant issue for people with diabetes. For the year 2022, CGMs had a total of 281 963 adverse events with 268 310 malfunctions, 13 644 injuries, and 9 deaths (as the manufacturer comments on each event, the total number of records was 583 321). The purpose of this article is to describe the most frequent CGM adverse events for both malfunctions and injuries for four different manufacturers.
To review the adverse event database, when a CGM user encounters a problem, they usually contact the manufacturer, who is required by US law to fill out US Food and Drug Administration (FDA) form 3500A to report the user’s complaint. Only 0.025% of complaints came directly from users. These results populate what is known as the MAUDE (Manufacturer and User Facility Device Experience) database. The database is searchable on the FDA website 2 but only returns 500 records per search and is difficult to use. Among the key fields in the database are the EVENT_TYPE (malfunction, injury, or death), BRAND_NAME (eg, DEXCOM G6), GENERIC_NAME (eg, CONTINUOUS GLUCOSE MONITOR), FOI_TEXT (text provided by the manufacturer to summarize the complaint or to comment on the event including testing results if the product was returned), and PROBLEM_CODE (a number chosen by the manufacturer which has an accompanying description). The field MDR_REPORT_KEY is a number used to combine tables. A typical event has the same MDR_REPORT_KEY for two or more records, with the FOI_TEXT field different for records with the same MDR_REPORT_KEY. As alluded to above, when several records have the same MDR_REPORT_KEY, one record is a description of the event, and one or more records are comments from the manufacturer about the event.
The US medical device reporting law (21CFR 803) states, 3 “If you are a device user facility, you must report deaths and serious injuries that a device has or may have caused or contributed to, establish and maintain adverse event files, and submit summary annual reports.”
“
(1) Failure,
(2) Malfunction,
(3) Improper or inadequate design,
(4) Manufacture,
(5) Labeling, or
(6) User error.”
Methods
Data were downloaded into an SQL Server database from the FDA website 4 for the year 2022. Queries were performed to obtain tables for four CGMs: Dexcom G6, all versions of Abbott Libre, Medtronic Guardian 3, and Senseonics Eversense. Each table’s record had a problem code (and definition), which is the manufacturer’s choice of the adverse event cause. Each of the four CGM products was further divided into two event types: malfunction and injury.
The eight resulting tables were sorted by descending frequency of the problem code. The problem code definitions were not always clear (or relevant to the complaint text)—note that problem codes were created for all medical devices, not just CGMs. Therefore, additional tables were created for each of the most frequent problem codes so that the text of the event (the FOI_TEXT field) could be examined. For example, for Dexcom G6, a top problem code was “no device output.” A table was created for Dexcom G6 malfunctions with this problem code and containing the FOI_TEXT field. Scrolling through these records while looking at the text in FOI_TEXT showed that virtually all records contained the term “transmitter failed error” in the description of the adverse event.
When scrolling through the records was insufficient to determine a unique cause, the data were downloaded into Excel and key terms were counted using a visual basic for applications program. If terms found accounted for at least 80% of the records, the terms found were used to describe the event.
For example, for a set of Dexcom G6 injury records, the problem code was “insufficient information.” In this case, examining the FOI_TEXT more closely, showed the cause was “skin reaction.” This is an example of the actual text.
IT WAS REPORTED THAT A SKIN REACTION OCCURRED. THE SENSOR WAS INSERTED INTO THE ARM ON (B)(6) 2022. ON (B)(6) 2022, THE PEDIATRIC PATIENT EXPERIENCED A SKIN REACTION WITH ITCHING AND INFECTION UNDER THE ENTIRE PATCH AREA. THE PATIENT CONSULTED A PHYSICIAN AND AN ORAL ANTIBIOTIC (FLUCLOXACILLIN 500 MILLIGRAM 4 TIMES A DAY) WAS PRESCRIBED. THE AREA WAS PREPARED WITH SOAP AND WATER BEFORE PLACING THE SENSOR ON THE BODY. AT THE TIME OF THE REPORT, THE PATIENT WAS DOING FINE. A PHOTOGRAPH WAS PROVIDED FOR INVESTIGATION. THE ALLEGATION WAS CONFIRMED VIA PHOTOGRAPHIC INSPECTION. THE PROBABLE CAUSE COULD NOT BE DETERMINED. NO ADDITIONAL EVENT OR PATIENT INFORMATION IS AVAILABLE.
Note, dates are often redacted by the FDA using the term (B)(6). For selected tables, all adverse event text was read.
Results
The results from the eight tables are shown in Tables 1 to 8.
Top six Dexcom G6 Malfunction.

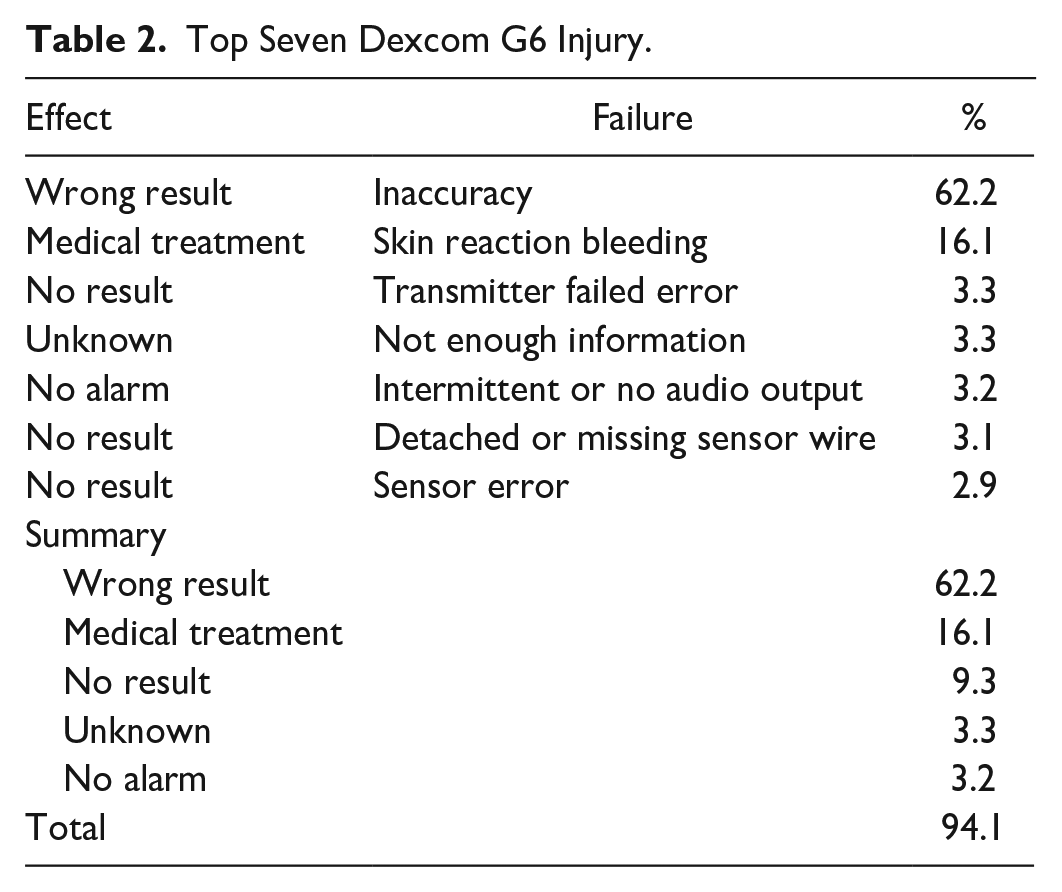
Top Seven Dexcom G6 Injury.
Top Two Abbott Libre Malfunction.

Top Five Abbott Libre Injury.

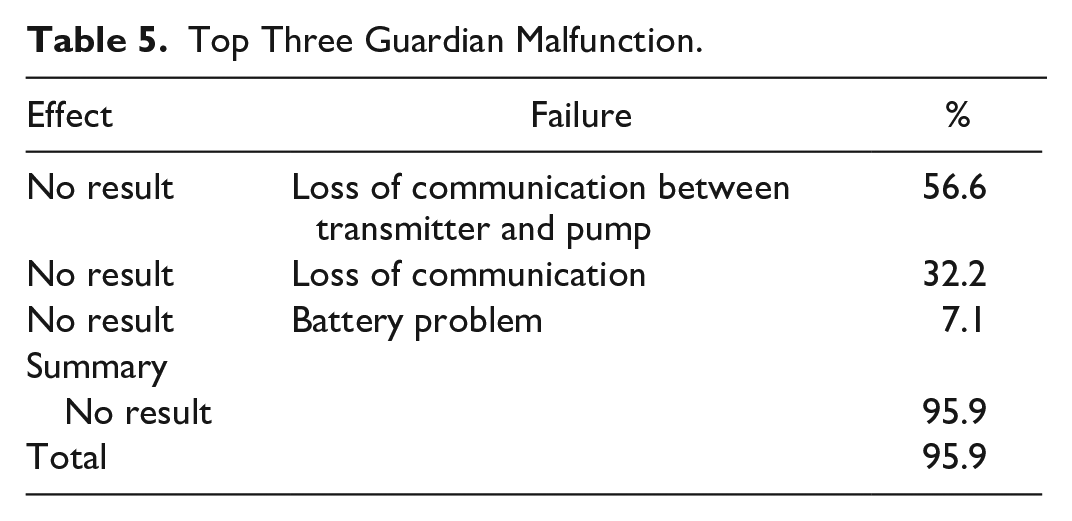
Top Three Guardian Malfunction.
Top Two Guardian Injury.

Top Three Eversense Malfunction.

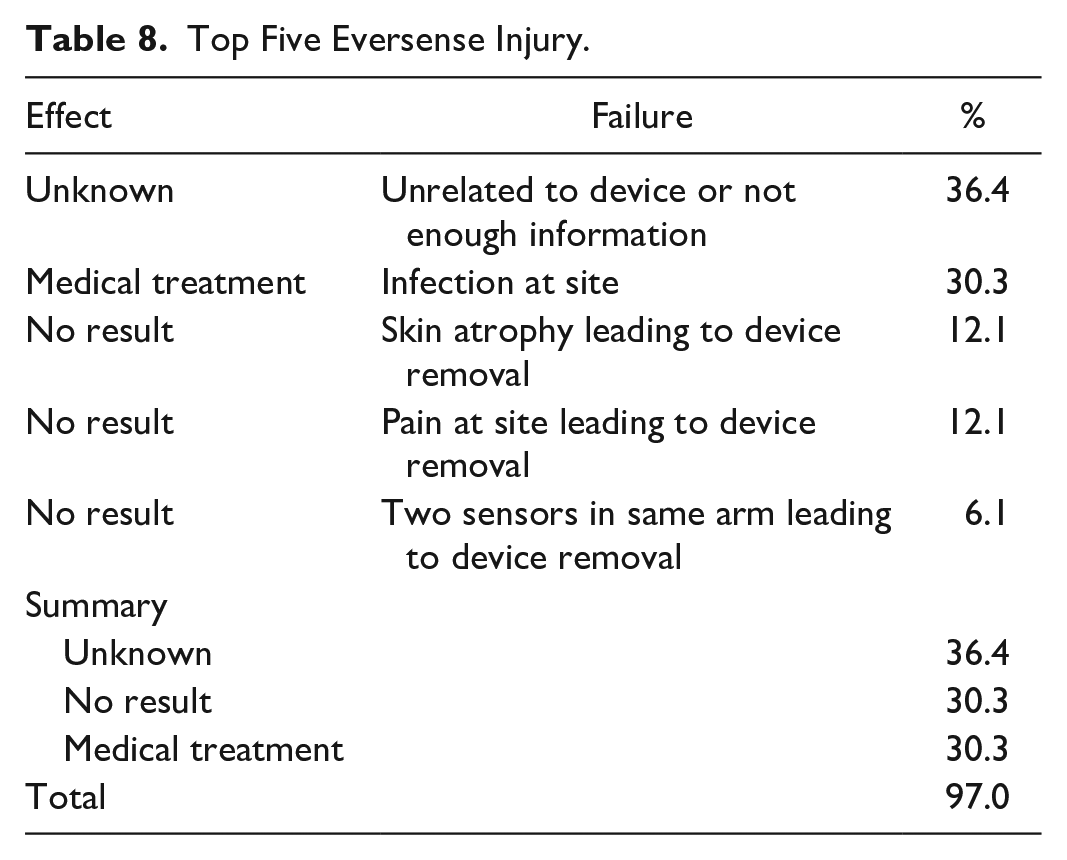
Top Five Eversense Injury.
Limitations
The CGM data did not always have uniform names for the field BRAND_NAME, and this field was sometimes blank. Also, a few adverse events were duplicated in the database. No attempt was made to deal with these problems. There is no guarantee that all adverse events were submitted or properly classified. MAUDE contains the description of the initial event. If a subsequent analysis of the event was conducted, it is unlikely to be entered in the MAUDE database. Thus, the “causes” listed in the above tables may merely be descriptions of events rather than true underlying causes.
Discussion
Manufacturers decide how to fill out Form 3500A including whether to classify an event as an injury or malfunction. Each manufacturer had a style in both assigning a problem code and how they filled out the text description for an adverse event. For example, Dexcom and Abbott used different problem codes and different text descriptions when the underlying complaint was inaccurate results. This difference in style could potentially also apply to classification of events. Although this was not studied, the following ratios (number of malfunctions/number of injuries) were observed: Eversense 0.6, Libre 1.7, DEXCOM G6 146.7, Guardian 196.9. The term inaccuracy is an unfortunate term because it implies that the CGM device is inaccurate, but user error cannot be excluded, nor can it be assumed that the comparison device (usually a blood glucose meter) is correct. But when users notice a large enough difference between the CGM and their glucose meter, they will use the term inaccuracy.
The safety and accuracy of a CGM device is assessed by the FDA. Their decision to accept for release for sale or reject a CGM amounts to deciding which is less harmful—harm caused by adverse events for a released product or harm caused by the lack of information due to the device having been rejected and not available. Clearly, CGM devices should be available. 1 But there are other ways of viewing acceptability; namely, how many adverse events are acceptable? The number of adverse events cannot be used to compare devices because as previously mentioned, one cannot be sure that all events were submitted or properly classified. And there are no usage numbers required for rates.
The number of CGM determinations in the US is the billions of glucose results. It is as if one poured billions of glucose results through a filter. What remains are the reported adverse events. Thus, a CGM adverse event is extremely rare if compared with the population of annual glucose results but less rare if compared with the number of US CGM users.
The FDA classification of events is based on outcomes. That is, if two CGM adverse event reports had a “no result” event and one did not result in an adverse outcome, it would be classified as a malfunction. If the outcome required medical intervention, it would be classified as an injury. This is different from FMEA (failure mode effects analysis) where classification is based on the severity of the potential outcome of the event, regardless of whether the outcome actually occurs. This distinction is important in quality improvement programs.
As previously reported, 5 no result is common adverse event occurrence and dominated these tables. Off label use (and misuse) were listed in comments by some manufacturers. Unfortunately, there was not enough information to reach any conclusions about user error.
In 2019, an article analyzed adverse events for two CGMs. 6 Other than that, there seems to be little interest in publishing about adverse events for CGMs. There are several potential reasons for this. For one, each year, there are roughly half a million records of CGM adverse events that are a challenge to analyze. The entries in the database are not always uniform. For example, there were 42 variations used for the Dexcom G6 product name.
Reducing the number of CGM adverse events requires the following steps: One must set an adverse event goal, collect adverse event data (in place from the FDA), analyze the data (eg, this article), propose ways to prevent the most frequent adverse events, and measure progress. A methodology for this has previously been reported, 7 and there is a well-known example for anesthesiology devices. 8
Conclusions
Creating tables based on manufacturer problem codes for their CGMs, followed by analysis of the adverse event text, facilitated the analysis of event causes. Otherwise, one would have to examine more than half a million records.
A common effect of several event causes prevented the user from obtaining a result. Inaccuracy was also a frequent complaint. Other causes were specific to that device. Analyzing adverse event data is the first step in trying to reduce the number of adverse events.
Footnotes
Abbreviations
CGM, continuous glucose monitor; FMEA, failure mode effects analysis; MAUDE, Manufacturer and User Facility Device Experience; FDA, US Food and Drug Administration.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.