
Abstract
Background:
For diabetes mellitus treatment plans, the consistency and quality of insulin drug products are crucial for patient well-being. Because biologic drugs, such as insulin, are complex heterogeneous products, the methods for drug product evaluation should be carefully validated for use. As such, these criteria are rigorously evaluated and monitored by national authorities. Consequently, reports that describe significantly lower insulin content than their label claims are a concern. This issue was raised by a past publication analyzing insulin drug products available in Canada, and, as a result, consumers and major patient organizations have requested clarification.
Methods:
To address these concerns, this study independently analyzed insulin drug products purchased from local Canadian pharmacies—including human insulin, insulin analogs, and porcine insulin—by compendial and noncompendial reversed-phase high-performance liquid chromatography (RP-HPLC) methods.
Results:
We demonstrated the importance of using methods fit for purpose when assessing insulin quality. In a preliminary screen, the expected insulin peak was seen in all products except two insulin analogs—insulin detemir and insulin degludec. Further investigation showed that this was not caused by low insulin content but insufficient solvent conditions, which demonstrated the necessity for methods to be adequately validated for product-specific use. When drug products were appropriately assessed for content using the validated type-specific compendial RP-HPLC methods for insulin quantitation, values agreed with the label claim content.
Conclusions:
Because insulin drug products are used daily by over a million Canadians, it is important that researchers and journals present data using methods fit for purpose and that readers evaluate such reports critically.
Background
The treatment for any form of diabetes mellitus (DM) is a complex and multifaceted process constantly adapting to new developments. In this area, it is important to be aware of the current quality of insulin drug products on the market. Because treatment for DM, particularly type 1 DM, often requires continuous parenteral (subcutaneous) insulin replacement therapy with recombinant human insulin (hI), analogs of hI, or animal insulin,1,2 product quality is impactful on patients' well-being.
For the regulation of biologic drugs including insulin products, national authorities such as Health Canada (HC), the U.S. Food and Drug Administration, and the European Medicines Agency are responsible for ensuring product access and quality.
In Canada, the evaluation and monitoring of biologic drugs are rigorous and data-driven. Drug submissions for market authorization in Canada require manufacturers to provide a comprehensive data package detailing drug quality, safety, and efficacy. 3 A part of this evaluation involves the thorough assessment of data from both long-term and accelerated stability studies conducted with appropriately validated methods. These methods and their validation are integral to ensuring that products will meet their label claim throughout the proposed expiry period. In Canada, further in-house laboratory testing of key analytical methods may be performed by HC to evaluate the suitability of proposed methods. Finally, in addition to laboratory evaluations, post-marketing surveillance, adverse event reporting, periodic quality reviews, and regular inspections of Good Manufacturing Practices (GMP) for drug storage facilities are conducted by HC to ensure continued product efficacy, safety, and quality.
In this context, it is important to fully understand and investigate reports that describe drug product deviations given the potential for patient impact. For example, in 2018, one such publication by Carter and Heinemann described finding lower amounts of active ingredients in marketed hI products than their label claim. 4 Using a mass spectrometry (MS) method originally developed for evaluating insulin content in plasma,5,6 it was concluded that no purchased product vial met the minimum insulin content specification of 95 U/mL, with some vial contents as low as 13.9 U/mL. 4 These observations were discussed by the authors as possibly caused by issues in the cold chain during transport, which suggested the widespread risk of potentially serious adverse events from product ineffectiveness. Understandably, this conclusion raised significant concerns from both health care providers and patients regarding product safety and efficacy. In response, numerous letters and publications from industry, academia, and patient groups were published to demonstrate the stability of insulin products and the effectiveness of the cold supply chain.7 -13 Notably, no major adverse events, clinical abnormalities, or product quality issues were identified in any routine regulatory surveillance from the United States, Canada, and Europe.
In this study, we demonstrate the importance of using methods fit for purpose when assessing insulin quality. Using pharmacopoeial methods routinely used to assess the quality of marketed insulin products in Canada, we show that the results are highly reliable and agree well with label claims. We also demonstrate how using methods not validated for specific products can produce unexpected results, which must be investigated further experimentally. Both the United States pharmacopoeia (USP29-NF24) and European pharmacopeia (EP, 01/2008:0854) reversed-phase high-performance liquid chromatography (RP-HPLC) methods were used to analyze several insulin drug products purchased from local pharmacies. First, a preliminary evaluation was performed for reference only, using the USP monograph for “Insulin.” This was an initial qualitative screen of the expected main insulin species in all products. Notably, two products, insulin detemir and insulin degludec, did not produce an insulin peak. A further analysis revealed that these chemically modified hI analogs required significantly higher nonpolar solvent conditions for successful elution. To demonstrate proper verification of the insulin content in drug products, a quantitative analysis was performed on products containing recombinant hI, porcine insulin, and one hI analog, using the corresponding insulin-type-specific USP (USP29-NF24) and EP monographs appropriately validated for quantitation. All products tested were within the accepted range of the label claim. Finally, these methods are compared to nonvalidated methods and discussed.
Methods
Chemicals
All chemicals were of analytical reagent grade. Acetonitrile, phosphoric acid solution (85%), and hydrochloric acid were purchased from EMD Millipore, Merck KGaA (Darmstadt, Germany). Ammonium bicarbonate, formic acid, sodium sulfate, and water graded for liquid chromatography-mass spectrometry (LC-MS) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Distilled water was deionized on a Nanopure Diamond system (Barnstead International, Dubuque, IA, USA).
Instrumentation
A Waters Alliance 2695 chromatograph equipped with a column heater and auto-sampler was used. A Waters 2998 UV-VIS photodiode array detector at 214 nm was used for detection (Waters, QC, Canada). Data acquisition and analysis was performed by using Empower 3 software (Waters, QC, Canada).
RP-HPLC Sample Preparation and Separation Conditions
For quantitation of insulin, samples were prepared and analyzed according to the appropriate type-specific USP (USP29-NF24) and EP (01/2008:0854) monographs. Where samples are not analyzed by type-specific monographs, a description of the method used is provided in text.
Monographs and column information (Table S1) for quantitative analysis are as follows: For USP analysis of hI products, the “Insulin Human Injection” monograph was followed and a Spherisorb ODS2, 150 mm × 4.6 mm, 3.0 μm particle size column (Waters, Milford, MA, USA) was used. For USP analysis of lispro products, the “Insulin Lispro Injection” monograph was followed, and a Spherisorb ODS2, 100 mm × 4.6 mm, 3.0-μm particle size column (Waters, Milford, MA, USA) was used. For EP analysis of hI, the “Insulin preparations, injectables” monograph was followed. For EP analysis of porcine insulin, the “Insulin, porcine” monograph was followed. For all EP monograph analyses, a Vydac C18, 250 mm × 4.6 mm, 5.0-μm particle size column (Hichrom Limited, Theale, Reading, UK) was used.
Chromatographic separations for all methods were carried out at 40°C with an isocratic elution requiring 40 minutes at 0.8 mL/min or 1.0 mL/min, depending on the monograph (Table S1). Eluent A was 0.2 M sodium sulfate, pH 2.3/acetonitrile (75%/25%).
For the analysis of insulin degludec and insulin detemir, the USP method for “insulin” was modified to include a gradient elution requiring 55 minutes of separation (Table S2) at 1.0 mL/min and 40°C. Eluent A was 0.2 M sodium sulfate, pH 2.3/acetonitrile (75%/25%), and eluent B was 0.2 M sodium sulfate, pH 2.3/acetonitrile (60%/40%).
Results
Preliminary Analysis of Canadian-Sourced Insulin Drug Products by a USP RP-HPLC Method for “Insulin”
To investigate the concerns raised by the Canadian public, 24 marketed insulin products (recombinant hI, hI analogs, and porcine insulin) were purchased from local pharmacies (Table 1). In an initial screening assessment, all insulin products were tested with the chromatographic procedure described in the RP-HPLC identification method of the USP monograph (USP29-NF24), “Insulin.” Although this chromatographic procedure is only intended for use with human and porcine insulin products, we used this as a rapid preliminary assessment to screen qualitatively for potentially abnormal insulin traces in all products. Indeed, most products—except for insulin detemir and insulin degludec—produced a single insulin peak, which was clearly visualized after 12 minutes and showed defined retention time shifts between products (Figure 1). Other peaks eluting prior to 12 minutes were identified as major excipients or other additives. Sample blank injections of each component showed no interference or co-elution with the main insulin peak (Figure S1), and expected excipients were separated for each product (Table 1). For appropriate identification and quantitation of hI analogs, further method validation would be required. However, these data were an encouraging indicator of quality in a wide range of Canadian-sourced products.
Purchased Marketed Insulin Products in Canada That Were Used in This Study.
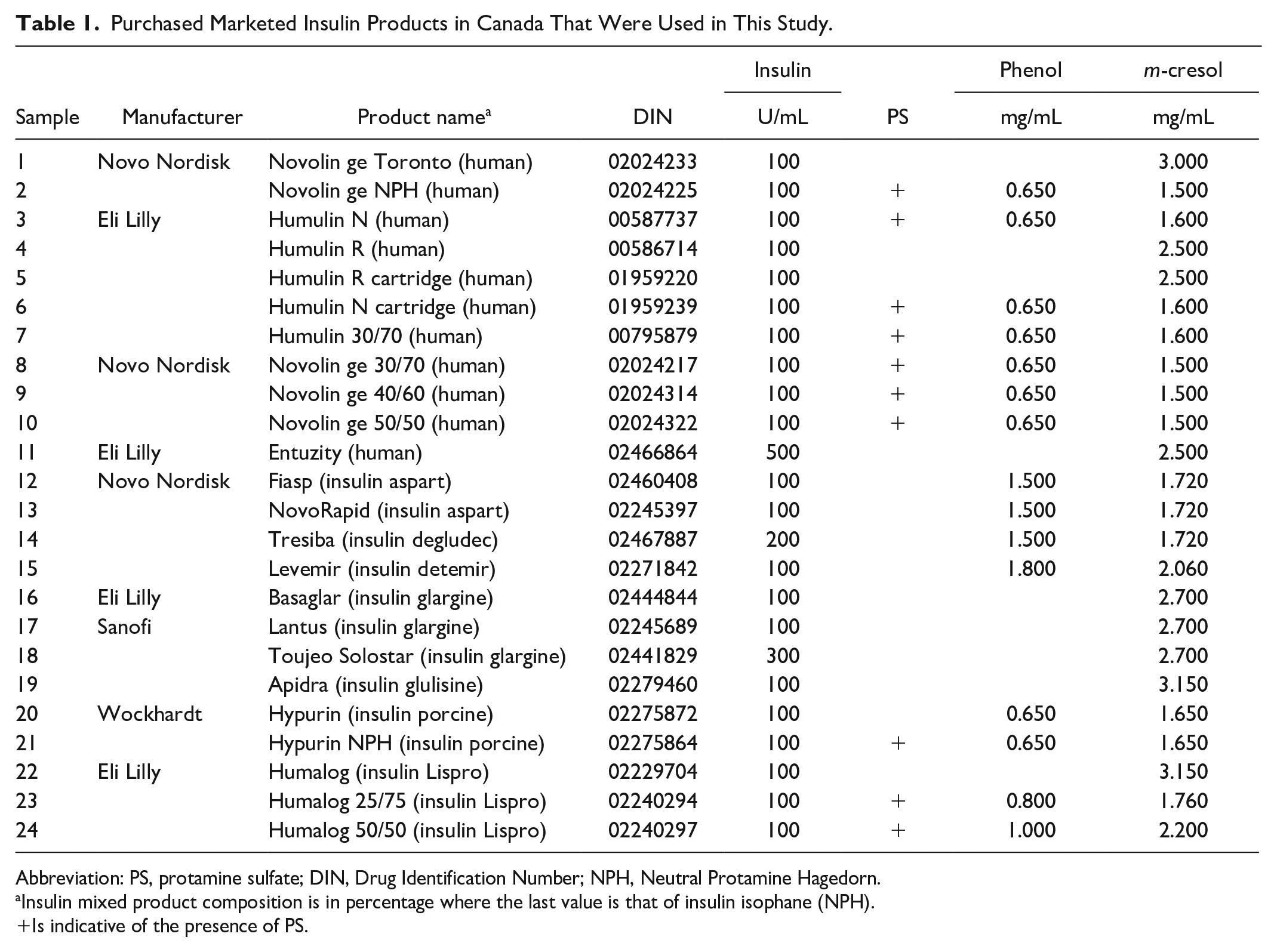
Abbreviation: PS, protamine sulfate; DIN, Drug Identification Number; NPH, Neutral Protamine Hagedorn.
Insulin mixed product composition is in percentage where the last value is that of insulin isophane (NPH).
Is indicative of the presence of PS.
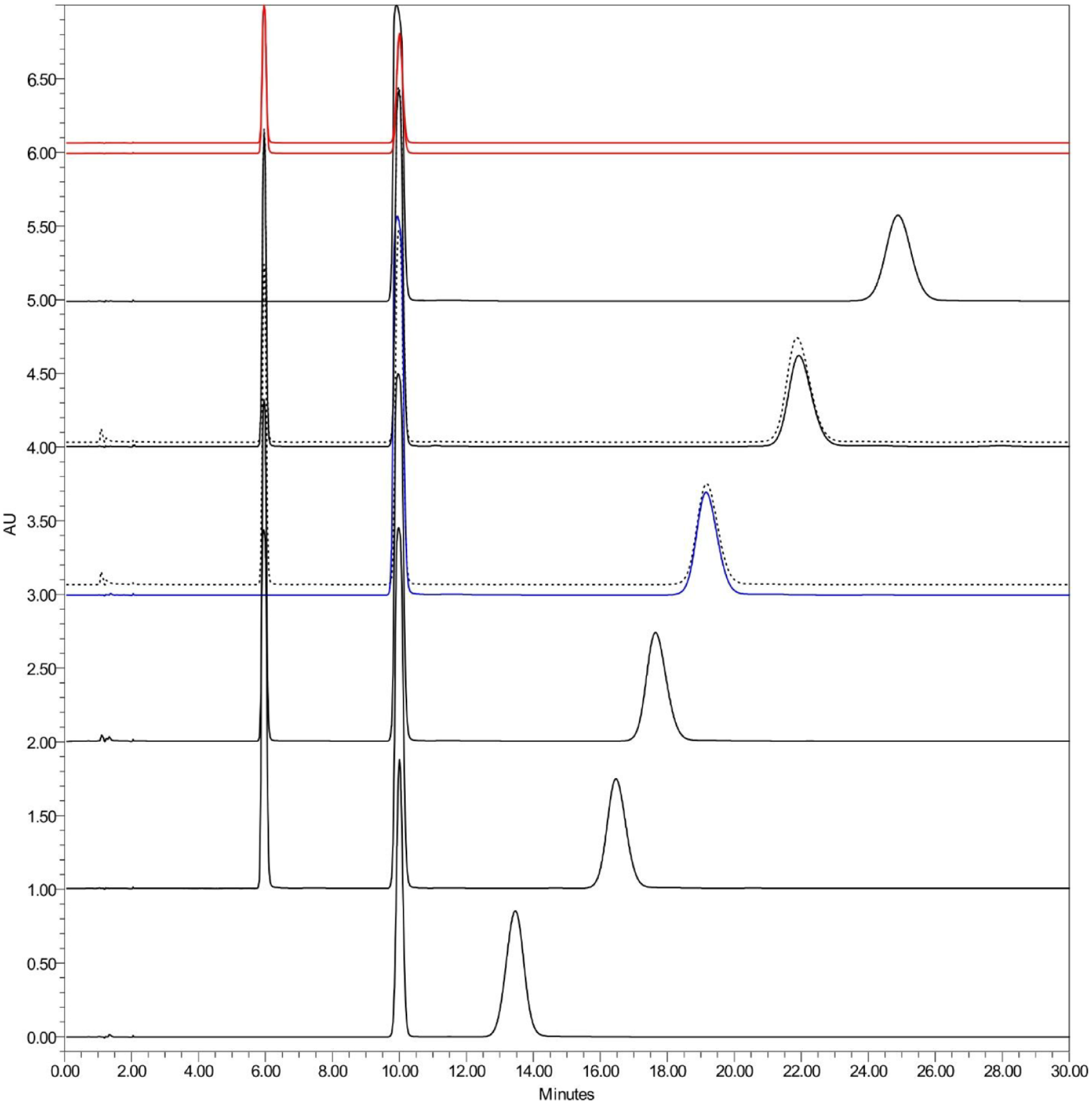
Representative traces of hI, porcine insulin, and hI analogs obtained by RP-HPLC using the USP monograph for “Insulin.” From bottom to top: insulin glargine, insulin aspart, insulin lispro, insulin (blue)/insulin NPH (blue, dashed), insulin porcine/insulin porcine NPH (dashed), insulin glulisine, insulin degludec (red), and insulin detemir (red) are displayed. Peaks prior to the insulin peak were identified as protamine sulfate (1.0 minutes), phenol (6.0 minutes), and meta-cresol (10.0 minutes), where present.
Modification of the USP RP-HPLC Method for Qualitative Evaluation of Chemically Modified hI Analogs
It was noted that no insulin peak was visualized for insulin degludec and insulin detemir (Figure 1, red tracings), which could be interpreted as lack of insulin content. However, these two hI analogs are chemically modified for increased hydrophobicity, which also suggests that the isocratic conditions of the compendial USP method were insufficient for elution. To investigate the elution behavior of these two products, the USP method for “Insulin” was modified to include a gradient toward higher organic solvent content (Table S2). Indeed, Figure 2 shows that elution of insulin detemir and insulin degludec required significantly more nonpolar conditions than all other products. Although this gradient method was not validated for insulin identity and quantitation, these data demonstrate the importance of confirming initial results with further experimentation and appropriate controls.
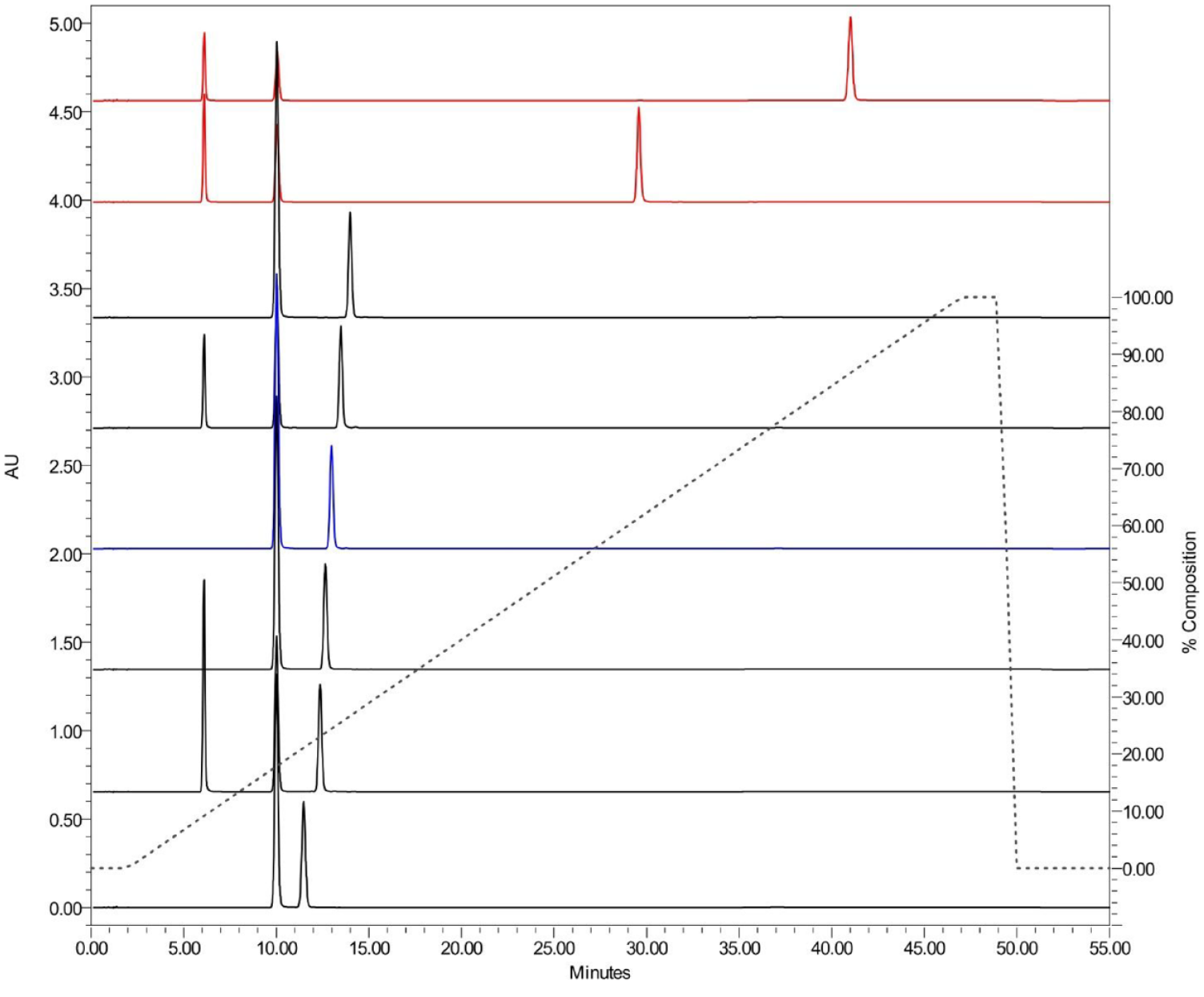
Representative traces of hI, porcine insulin, and hI analogs obtained by a gradient separation included in the USP monograph for “Insulin.” From bottom to top: insulin glargine, insulin aspart, insulin lispro, insulin (blue), insulin porcine, insulin glulisine, insulin degludec (red), and insulin detemir (red). Peaks prior to the insulin peak were identified as protamine sulfate (1.0 minutes), phenol (6.0 minutes), and meta-cresol (10.0 minutes), where present. Abbreviations: hI, human insulin; USP, United States pharmacopoeia.
USP and EP RP-HPLC Methods for Quantitation of Insulin Content
To properly verify that insulin content in marketed drug products met label claims, a quantitative analysis was performed on a subset of purchased products using the RP-HPLC method from the corresponding type-specific USP (USP29-NF24) and EP (01/2008:0854) monographs. Insulin content was quantitated in 11 recombinant hI products, two porcine insulin products, and three insulin lispro products (Table 2). Accurate insulin quantitation was achieved by comparison to the appropriate compendial-specific certified reference standard (Table S3) prepared according to the respective monograph detailed in the Materials and Methods section.
Concentration of Insulin in Samples Using USP and EP Monograph.
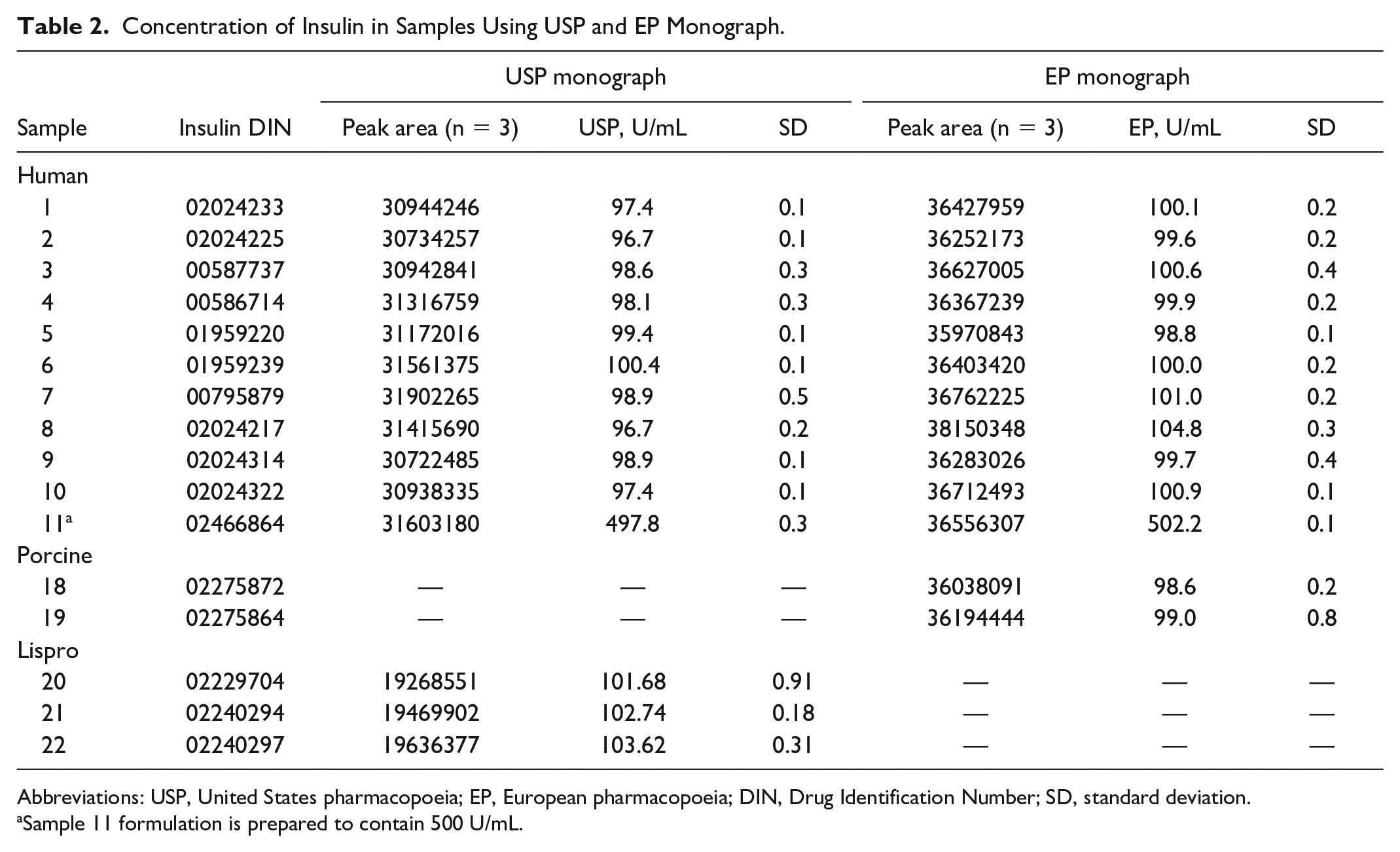
Abbreviations: USP, United States pharmacopoeia; EP, European pharmacopoeia; DIN, Drug Identification Number; SD, standard deviation.
Sample 11 formulation is prepared to contain 500 U/mL.
The resulting calculated measurements (Table 2) showed that insulin content in these tested products ranged between 96.7 U/mL and 104.8 U/mL, which met the potency criteria (100.0 ± 5.0 U/mL). Sample 11 tested in this study was formulated at 500 U/mL, which was measured at a range between 497.8 U/mL and 502.2 U/mL. In addition, the results between both USP and EP monographs were highly comparable for content assessment in hI products. In summary, the insulin content values of all tested products met the potency criteria outlined in all compendial monographs.
Conclusions
In this study, we used a compendial RP-HPLC method to demonstrate that insulin content across Canadian marketed drug products was highly consistent and within the accepted range of the stated label claim. In contrast, findings by Carter and Heinemann reported significant loss in the active ingredient of insulin drug vials (>50%). 4 Upon review of this report, it appeared that method details were limited and that the MS method used to assess insulin content was not validated as a quantitative method for marketed drug products. Most parameters were referenced from an LC-MS/MS method, originally developed by Chambers et al,5,6 for the simultaneous differentiation and quantitation of hI and its analogs in human plasma samples. This original LC-MS/MS approach required an extensive sample cleanup protocol, critical for a plasma matrix, and the analysis was performed on an ultra-performance liquid chromatography (UPLC) system coupled to a triple-quadrupole mass spectrometer, with separation carried out on a Charged Surface Hybrid (CSH) C18 (2.1 mm × 50 mm) column. In comparison, the adapted MS method by Carter and Heinemann did not provide details on sample preparation, and the analysis was performed on a quadrupole time-of-flight mass spectrometer, with separation carried out on a Cortecs C18 (2.1 mm × 30 mm) column. 4
Given the differing parameters and missing details, some questions on method transferability were raised. For example, when making changes to the column, the general EP monograph for chromatographic separation techniques recommends maintaining parameters such as the length to particle size ratio and the physicochemical characteristics of the stationary phase. Gradient systems require greater caution than isocratic systems. It would be good practice to show that these changing parameters did not impact quantitative measurements. In addition, past work 13 demonstrated that the solid phase extraction step from Chambers et al 5 disproportionately reduced the recovery of hI compared to bovine insulin, when using the Cortecs C18 column. However, when the Solid-Phase Extraction (SPE) step was removed, LC-MS analysis produced comparable results to the USP method.
Thus, while Carter and Heinemann proposed a breakdown in the cold chain supply as an explanation for their observations, limitations of the method could also explain the discrepancy. 4 Because the original study did not detail sample preparation steps relevant to an applicable matrix and method validation was not performed, it is difficult to evaluate the root cause. These limitations have been extensively discussed elsewhere.7,8 As shown in this study, even compendial methods must be validated for use with specific product types. Notably, the results of our current study agree with other reports that confirmed adequate insulin concentrations in marketed products around the world. 14 These were conducted with a wide range of methodologies, including RP-HPLC (compendial), 9 RP-UPLC, 10 Nuclear Magnetic Resonance (NMR), 11 MS, 12 and LC-MS. 13
The work herein emphasizes the importance of using appropriate and well-controlled methods to assess quality specifications of biological products. Reports of novel analytical approaches without a comparison to compendial monograph methods should be carefully evaluated if proper method qualification and validation data are not provided.
Supplemental Material
sj-docx-1-dst-10.1177_19322968231159360 – Supplemental material for Regulatory Verification by Health Canada of Content in Recombinant Human Insulin, Human Insulin Analog, and Porcine Insulin Drug Products in the Canadian Market Using Validated Pharmacopoeial Methods Over Nonvalidated Approaches
Supplemental material, sj-docx-1-dst-10.1177_19322968231159360 for Regulatory Verification by Health Canada of Content in Recombinant Human Insulin, Human Insulin Analog, and Porcine Insulin Drug Products in the Canadian Market Using Validated Pharmacopoeial Methods Over Nonvalidated Approaches by Barry Lorbetskie, Stewart Bigelow, Lisa Walrond, Agnes V. Klein, Shih-Miin Loo, Nancy Green, Michael Rosu-Myles, Xu Zhang, Huixin Lu, Michel Girard and Simon Sauvé in Journal of Diabetes Science and Technology
Footnotes
Acknowledgements
The authors thank Drs. Roger Tam, Yi-min She, and Michael Johnston (Health Canada) for their critical review of this manuscript.
Abbreviations
DM, diabetes mellitus; EMA, European Medicines Agency; EP, European pharmacopoeia; HC, Health Canada; hI, human insulin; MS, mass spectrometry; RP-HPLC, reversed-phase high-performance liquid chromatography; UPLC, ultra-performance liquid chromatography; USFDA, US Food and Drug Administration; USP, United States pharmacopoeia.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Health Canada.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.