
Abstract
One of the most prevalent complications of diabetes mellitus are diabetic foot ulcers (DFU). Diabetic foot ulcers represent a complex condition placing individuals at-risk for major lower extremity amputations and are an independent predictor of patient mortality. DFU heal poorly when standard of care therapy is applied. In fact, wound healing occurs only approximately 30% within 12 weeks and only 45% regardless of time when standard of care is utilized. Similarly, diabetic foot infections occur in half of all DFU and conventional microbiologic cultures can take several days to process before a result is known. DFU represent a significant challenge in this regard because DFU often demonstrate polymicrobial growth, become resistant to preferred antibiotic therapy, and do not inform providers about long-term prognosis. In addition, conventional culture yields may be affected by the timing of antibiotic administration and collection of tissue for analysis. This may lead to suboptimal antibiotic administration or debilitating amputations. The microbiome of DFU is a new frontier to better understand the interactions between host organisms and pathogenic ones. Newer molecular techniques are readily available to assist in analyzing the constituency of the microbiome of DFU. These emerging techniques have already been used to study the microbiome of DFU and have clinical implications that may alter standard of care practice in the near future. Here emerging molecular techniques that can provide clinicians with rapid DFU-related-information and help prognosticate outcomes in this vulnerable patient population are presented.
Keywords
Introduction
Diabetes mellitus (DM) may affect every third adult American by 2050 1 and up to 35% will develop diabetic foot ulcers (DFU). 2 Trends indicate diabetes-related non-traumatic lower extremity amputations (LEA) are on the rise again. 3 This trend is strongly influenced by the presence of DFU and LEA remain one of the most devastating complications of DM. Even when standard of care is met, including wound debridement, dressing changes, pressure offloading, and diabetes control, less than half of DFU will heal and up to 60% will develop into diabetic foot infection (DFI).4–6 Therefore, factors other than those addressed by standard of care management could be important to better understand and improve treatment for DFU.
One such area that has undergone intense investigation within the past decade is the human microbiome. Broadly, the human microbiome represents the microorganisms that reside on or within human tissues and may include the genes and genomes of members of the microbiota. 7 Scientific discovery over the past decade has revealed an unappreciated complexity to skin microbiome, including wound healing. In the skin, commensal bacterium induce cutaneous interferon and interleukin IL-17A producing T-cell release. 8 When commensal-specific T-cell responses are activated by a skin disturbance (such as DFU), its response alters signaling to microbial communities. 9
This signaling disturbance may affect the diabetic foot because the pedal skin microbiome has reduced diversity. 10 Additionally, the diversity of commensal bacterium responds dynamically to the microenvironment, including antibiotic use. 11 In fact, increased microbial diversity has been positively correlated to DFU healing12,13 whereas overabundance of specific microbiota diminishes the migration and differentiation of mesenchymal stromal cells. 14 Thus, developing a more complete understanding of the microbiome in DFU is critical to enhance therapy. However, conventional culture, where a biospecimen is sent for microbiological analysis, may not fully represent host–microbiome interaction.
Instead, emerging molecular technology tools offer improved granularity and specificity to study the organisms of the DFU microbiome. The shift toward these dynamic culture-independent methods may improve our understanding of the relationship between the organisms in the DFU microbiome, patient outcomes, and wound progressions.
The Diabetic Foot Microbiome
Literature on conventional culture techniques demonstrate significant DFU microorganism heterogeneity.15,16 The organisms most commonly cultured are reflective of soft tissue infections. Organisms include Staphylococcus aureus (SA), methicillin-resistant Staphylococcus aureus (MRSA), Group B Streptococcus, Enterococcus faecalis, Enterococcus faecium, and coagulase-negative staphylococci15–18 among others, which have been discovered because of emerging technology use (Figure 1). Not unexpectedly, the diversity of the microbial constituency in DFI is broad, polymicrobial, and mimics diabetic skin. 19
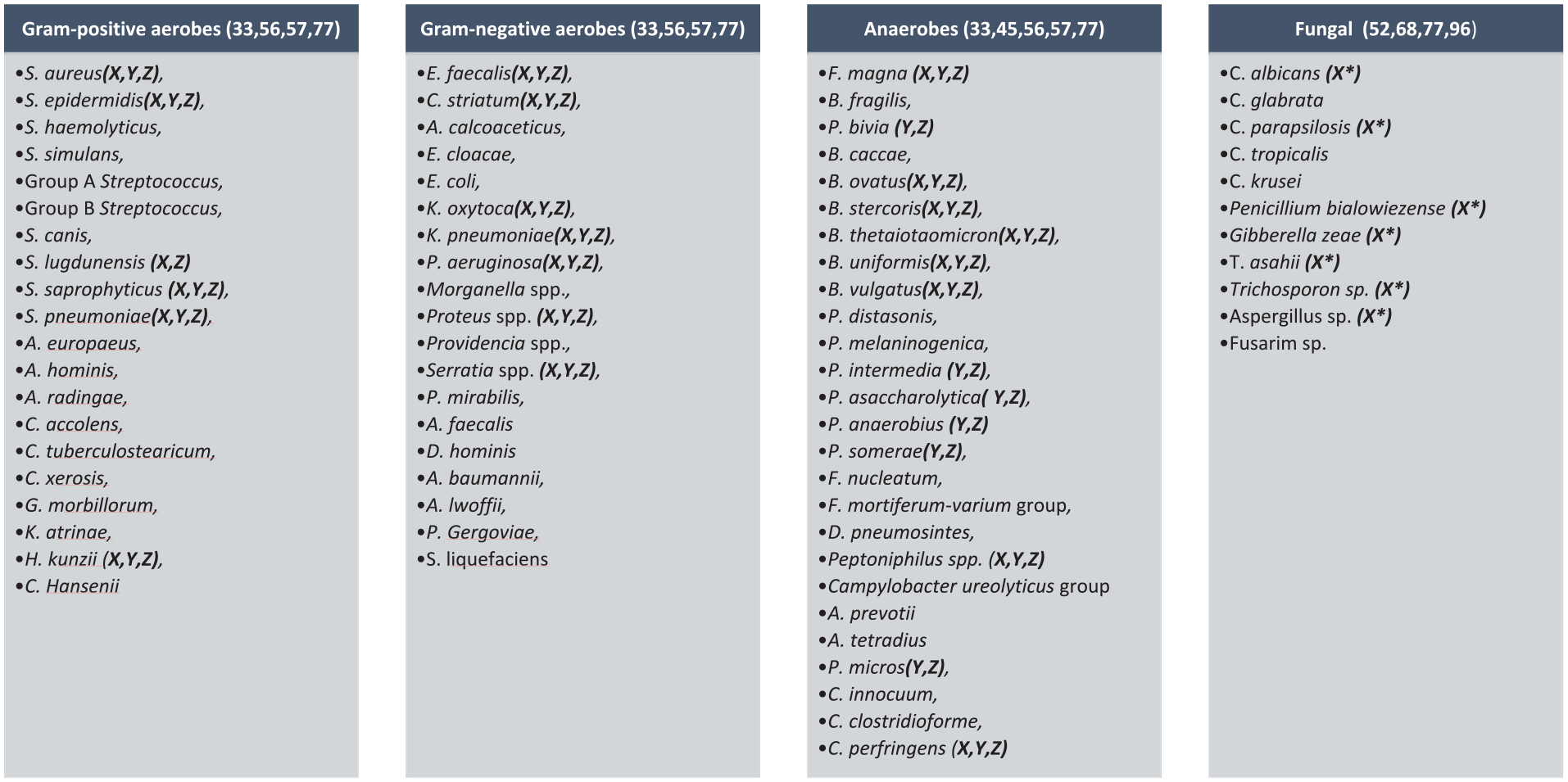
Conventional Culture Microbial Constituency: Using conventional culture in DFU, a wide and diverse microorganism population has been discovered. Uncommonly cultured organisms identified using 16S rRNA (X), sequencing of the nuclear ribosomal internal transcribed spacer 1 (ITS1; X*), metagenomics (Y), and culturomics/MALDI-TOF-MS (Z) highlight in specific the presence and previously underreported abundances of anaerobic and fungi in the DFU microbiome.
Current Methods to Investigate Bacterial Presence
When DFI is identified, standard of care includes conventional culture. The International Working Group on Diabetic Foot and Infectious Disease Society of America diabetic foot guidelines recommend soft tissue cultures rather than wound swabs.17,20 Tissue cultures have high concordance at all DFU depths while wound swabs become discordant with increased DFU depth. 21 If only a wound swab can be performed, it should be completed after wound debridement.22,23
After processing, clinical isolates are subject to antibiotic sensitivity testing. Techniques include broth micro-dilution, antimicrobial gradient methods, disc diffusion, and are available in various commercial systems. Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines provide standardization for processing.24,25 After isolate identification, an antibiotic minimum inhibitory concentration is determined to eradicate the microbe. Infectious disease (ID) specialists then narrow antibiotic therapy according to culture results and this results in improved outcome data. 26
The use of conventional culture is not without its limitation, the most important of which is time. When a patient has a DFI, the patient will be started on empiric therapy until final cultures allow for appropriate narrowing of antibiotic regimen. 17 Historically, awaiting the final culture can delay optimal antimicrobial stewardship practices by several days or longer if awaiting fungal and acid-fast bacilli cultures. Together, a potential for prolonged hospitalization and unnecessary antibiotic administration may place a patient at risk to develop multidrug resistant organisms (MDRO).27-31
Advanced Methods for Microbiome Analysis
Molecular techniques offer promise in rapidly differentiating commensal and pathogenic organisms and include:
Polymerase chain reaction (PCR)
Microarray technology
Next (second) generation sequencing—16S ribosomal RNA (16S rRNA)
Matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS)
Whole genome sequencing (WGS) including third generation sequencing.
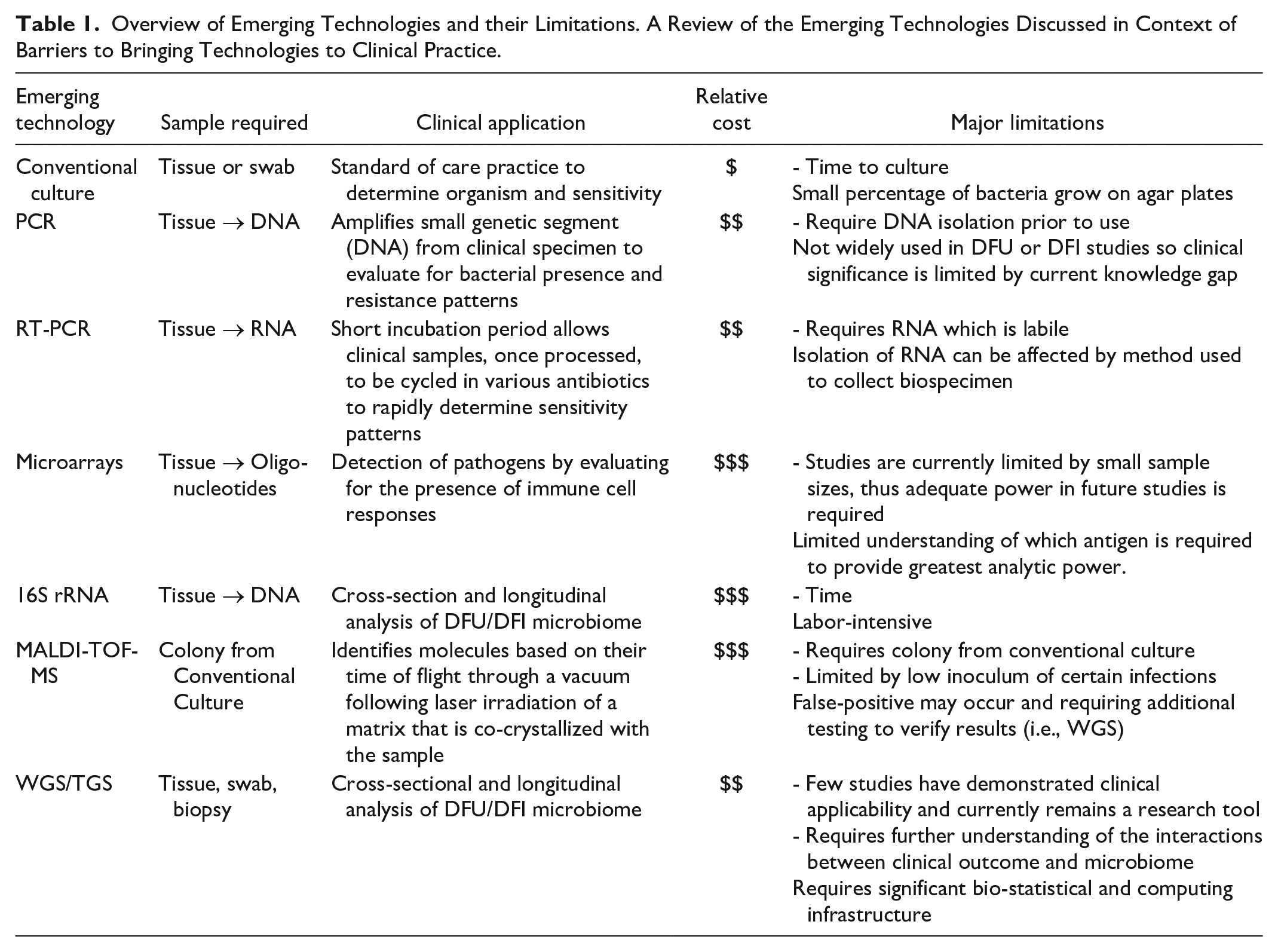
Each has contributed significantly to the understanding of the diabetic foot microbiome but each has limitations (Table 1). The techniques are highlighted and clinically relevant data is provided in each subsection below.
Overview of Emerging Technologies and their Limitations. A Review of the Emerging Technologies Discussed in Context of Barriers to Bringing Technologies to Clinical Practice.
PCR
Amplification of sequence-specific nucleic acid information is used in microbiologic pursuits to determine specific pathogens. PCR is essential in next generation sequencing and increases 16S rRNA feasibility (Figure 2). PCR amplifies a small genetic (DNA) segment processed from clinical specimens (biopsy, swab, tissue), as opposed to the whole genome, and analysis of the amplified segment can be assessed for known pathogenic genotypes. This requires DNA isolation from the clinical sample before PCR testing can occur.
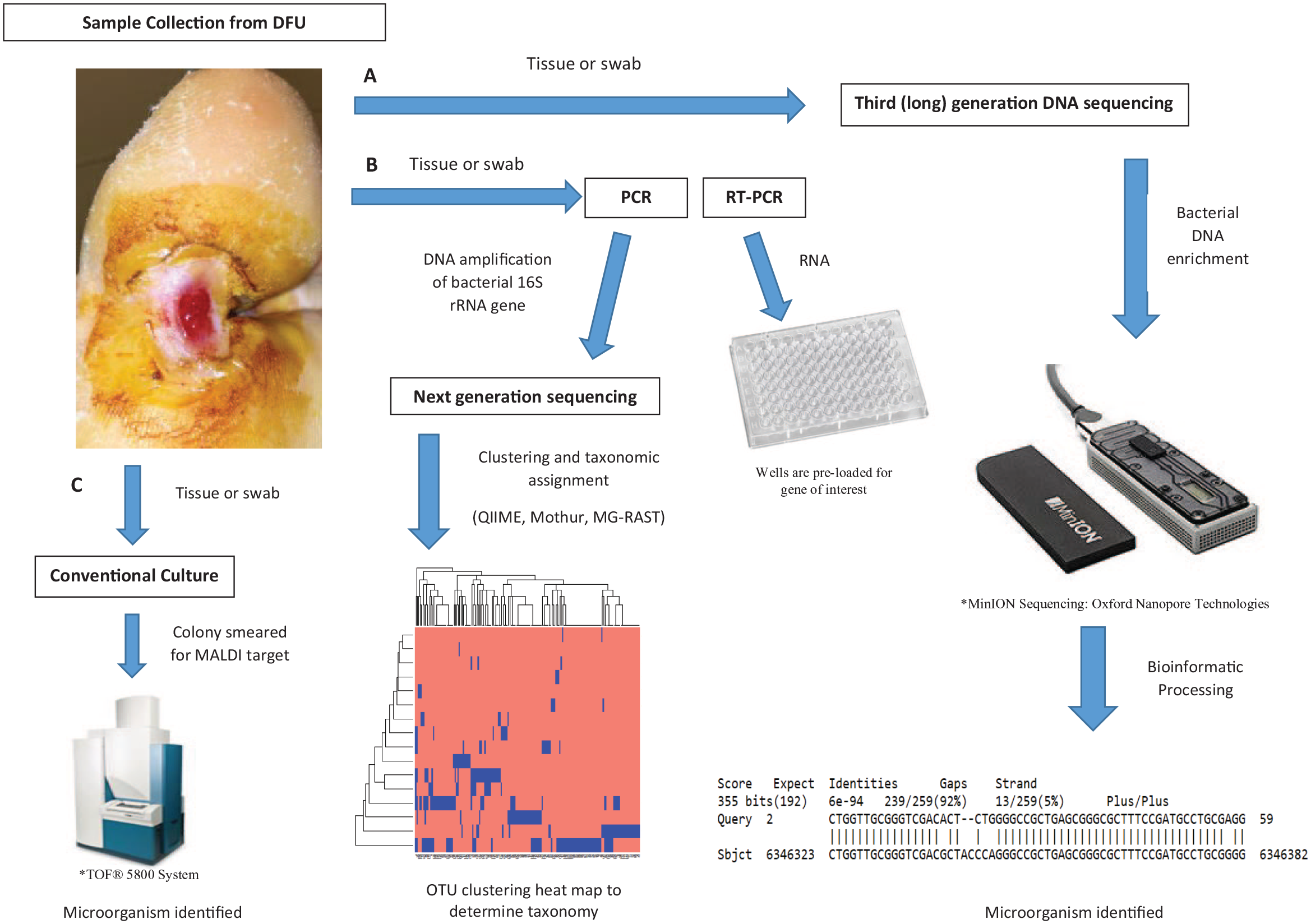
Comparative process between DFU sample collection and genomic evaluation via various modalities. (A) Third generation sequencing, (B) Next generation sequencing, and (C) culturomics/MALDI-TOF-MS are available to identify microorganism(s) from DFU biospecimen.
The classic example is identification of MRSA mecA gene. MRSA is present in approximately 10–32% of DFI and is associated with a higher rate of treatment failure, morbidity, and hospitalization cost.32,33 The mecA gene encodes a modified penicillin-binding protein with reduced affinity for β-lactam antibiotics and is widely used in commercial systems for identification purposes. Clinical samples can be used and processed to identify the presence of MRSA (mecA) in under 2 hours.32,34 When implemented clinically, specifically for mecA gene presence, PCR improves correct antibiotic administration by an average of 25.4 hours in patients with bacteremia. 35 PCR is also useful when it is not prudent to discontinue antibiotic therapy because it is able to diagnose unusual (difficult to culture) pathogens.36–38
PCR can be used in the identification of bacterial resistance. Enterococcus faecium, SA, Klebsiella pneumonia, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp (ESKAPE pathogens) are common in DFI and are often resistant when cultured. 39 PCR assays for identifying cephalosporinase- and cabapenemase-encoding genes in these bacteria are known and include KPC, NDM, IMP, VIM, AmpC, SHV, and OXA.40-44 However, PCR use in DFI is not commonly performed, demonstrating a gap in care for these individuals that should be explored in future studies.
When PCR tests are used elsewhere on the body, it can have utility in DFI management. It is well recognized the nares can determine MRSA colonizer status. When evaluated in the context of a DFI, a positive MRSA nasal swab is not predictive of causative organism, but a negative MRSA nasal swab rules out MRSA-colonizer status with 90% accuracy. 45 Thus, the incorporation of PCR testing to evaluate the host’s microbiome has utility and may promote antimicrobial stewardship in DFI.
Another such organism known to receive a great deal of attention in DFI is Pseudomonas. Pseudomonas is common in puncture wound osteomyelitis, in intravenous drug users, 46 and in patients who use an active wound dressing that promotes a moist wound environment. 47 In spite of evidence that Pseudomonas is an uncommon isolate in DFI48,49 empiric coverage of Pseudomonas is still provided broadly. One study demonstrated despite adherence to best national practice guidelines in Australia, 78% of DFI patients received anti-pseudomonal therapy even though only 8% of cultures had positive growth for Pseudomonas. 50 Very few publications comment on DFI antibiotic stewardship49,51 and there is growing concern for resistance to preferred treatment. 39 This excessive use of broad-spectrum antibiotics can be alleviated by rapid bacterial identification. Current open array technologies allow for the creation of a tailorable PCR test, up to 3,000 real-time reaction plates, which would ensure immediate judicious antibiotic use and avoids the lag time, which can be days, associated with conventional culture and sensitivity data. A challenge related with the use of a tailorable PCR test is understanding the association between phenotypic traits and microbiome parameters (bacterial type and abundances). These associations are not well-defined and characterizing these relationships are an important step to create a PCR test which is cost-effective and utilitarian in DFI management.
PCR can quantify the number of gene copies per given sample. When combined with the shorter incubation of real-time PCR (RT-PCR), rapid identification of sensitive- and resistant- organisms is possible. In fact, RT-PCR allows for bacteria to be cycled in various antibiotics and evaluated for resistance.52,53 The limitations of RT-PCR is, unlike traditional PCR which measure the determinants of resistance (i.e. mecA gene) using DNA, RT-PCR requires use of RNA. RNA is extremely labile compared to DNA. Thus, clinical samples present special concerns because inconsistencies in sample collection and processing can lead to variable RNA quality 54 and affect testing results.
Microarrays
Microarrays are similar to PCR in that they assess a specific sequence of nucleic acid. Microarrays use complementary oligonucleotides to identify a sequence of interest. Antibiotic resistance assays are commercially available for multiple antibiotic drug classes and use of microarrays have shown feasibility to distinguish between noninfected and infected DFU based on relative quantities of virulent genes. 55
One such example is a multiplex immunoassay of IgG in serum and medium-enriched for newly synthesized anti-SA antibodies (MENSA). It was developed from stimulated circulating plasmablasts. 56 Historically, SA was thought to be involved in 50% of DFI20,57 but evidence using methods described herewith in suggest an increasing role of polymicrobial and anaerobic-dominated infection. 58 However, the use of the immunoassay resulted in broad applicability of serum and MENSA for diagnosis of SA DFI and longitudinal tracking of MENSA was predictive of DFI outcomes. 56 This finding mirrored more morbid outcomes following prosthetic joint infections when SA antibodies against IsdA, IsdB, and IsdH were present. 59 The immunoassay also determined that some antigens confer protective effects in DFI.60,61 Caution is warranted as current knowledge of antigens involved in DFI is incomplete.
Importantly, oligonucleotide-based microarrays may suffer reduced sensitivity in the polymicrobial DFI environment. 56 Distinguishing between the infecting organisms and outcome prognostication is essential to guide future therapies and more comprehensive studies are needed.
16S rRNA
The most commonly employed technique to investigate the DFU microbiome is 16S rRNA analysis. It leverages the highly conserved 16S rRNA gene, present in all DNA lifeforms, and investigates the gene’s nine hypervariable regions to identify diversity between bacterial taxa. 62
The initial DFU study investigating bacterial constituency used partial ribosomal amplification and pyrosequencing, full ribosomal amplification, cloning, and Sanger sequencing, density gel electrophoresis and identified species based on operational taxonomic units (OTU). 63 Subsequent studies used clinical techniques including wound swabs, tissue debridement (sharp/surgical debridement), and curettage to procure tissue for DFU microbiome investigations. They have demonstrated each DFU is highly diverse and has quantities of bacteria previously not appreciated through conventional culture techniques (Table 2).
16S rRNA Analysis of Non-Infected DFU Microbiome. A Review of Pertinent Studies Using 16S rRNA to Study the Microbiome and Highlighting the Sample Size and Culture Techniques used in Study Design.

Microbiome communities are prognostic of outcome. Sloan et al. demonstrated bacterial communities with a dominance of Enterobacteriaceae and strict anaerobes have worse outcomes. 12 DFU microbiota were found to exist in one of four community types and experienced frequent and nonrandom transitions. 11 Antibiotics may destabilize the microbiome instead of altering diversity or increasing relative abundance of specific taxa. 11 Specific biofilms from SA and Pseudomonas are shown to diminish the migration ability of cells and hinder multi-lineage differentiation of mesenchymal stromal cells (MSC). 14 Furthermore, fungal prevalence in DFUs was previously underestimated by conventional culture.68,69
16S rRNA has also demonstrated the microbiome response following surgical intervention. When surgery on an infected foot is performed, marked increases in abundances of Actinomycetales and Staphylococceae correlate with DFU healing. In contrast, non-healing DFU have a higher abundance of Bacteroidales and Streptococcaceae spp. following treatment. 65
MALDI-TOF-MS
MALDI-TOF-MS identifies molecules based on their time of flight through a vacuum following laser irradiation of a matrix that is co-crystallized with the sample. The time of flight allows for determination of the mass/charge ratio of the ions present and a spectrum of the sample is generated. The analyte spectrum is then compared to a reference database.
The use of MALDI-TOF-MS has reduced time-to-organism-identification and time-to-effective-and-optimal-antibiotic-therapy. A hospital intensive care unit was able to reduce mortality and hospitalization by 6 days with MALDI-TOF-MS. 70 In DFI, MALDI-TOF-MS has broad applicability and is cost-effective with expenses of $1.00 per tested isolate. 71 MALDI-TOF-MS has identified rare organisms causing osteomyelitis71,72 and is accurate in species recognition including anaerobes, which can be difficult to isolate without special techniques. 73
Although MALDI-TOF-MS can determine speciation, several concerns arise, making it less feasible to use in a clinical setting. First, the quantity of microorganisms in different tissue types may be problematic. In bone and joint infections, MALDI-TOF-MS is limited because of low inoculum. 74 Another limitation is organism resistance patterns, which may have similar curves. For example, in clinical isolates of Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa no reliable peak pattern identifies β-lactamase resistance. 75 Finally, considerable logistical planning is required to incorporate MALDI-TOF-MS once conventional culture colonies are visualized to ensure timeliness of the results.
Additionally, adopters MALDI-TOF-MS may need to perform additional tests to ensure accuracy. A case report identifying a rare soil organism causing DFO had been identified as Clostridium sphenoides by MALDI-TOF-MS, but as Clostridium celerecrescens using 16S rRNA. 71 When discordance manifested in this case, the authors suggested using whole genome sequencing (WGS) to distinguish discrepancies. 71
Whole Genome Sequencing
MALDI-TOF-MS and 16S rRNA sequencing platforms are not immune to false positives. Whole-genome sequencing (WGS) can be used to differentiate conflicts between MALDI-TOF-MS and 16S rRNA sequencing in the microbiome.
Metagenomics information is vast. Independently sequencing smaller fragments of DNA is the basis of WGS. MRSA WGS, for example, lead to an understanding why SA is able to infect humans of diverse genetic backgrounds, eliciting severe immune reactions, and has over 70 candidate virulence factors. 76
Open source resources such as QIIME, Mothur, and MG-RAST afford investigators an ability to focus on three importance vectors of chronic wounds: microbial load, microbial diversity, and the presence of pathogens. 77 By using WGS, investigators can identify all genetic material in a wound: bacterial, viral, and fungi. For example, culture independent methods have demonstrated Candida spp. and Cladosporidium spp. to be common in DFU 69 and is consistent with surgical literature but went previously underreported.68,78 Leveraging WGS to understand the infinite interactions between multiple kingdoms in DFU may guide therapy and create personalized medicine depending on which pathogens are identified and dominant.
To increase the utility of WGS, additional navigation of sequence nuance is required. Distinction between living (host) cell DNA and dead (host) cell DNA is difficult; approximately 90% of DNA from a sample is human. 79 In addition, bacterial antibiotic resistance genes are ubiquitous in WGS as a result of contamination with DNA from other microorganisms and the host. 80 Furthermore, the ability to perform WGS across institutions varies. In its current form, WGS remains a bench oriented test—it is prohibitively expensive as compared to conventional culture and is not routinely performed in clinical laboratories. This is largely because WGS lacks automation and data interpretation requires a biostatistician which restricts clinical applications. 81
Third Generation Sequencing (TGS)
Third generation sequencing evaluates complete nucleotide sequences. Unlike 16S rRNA, because the strands of DNA are not separated into smaller segments, there is no need for PCR amplification and synthesis. By eliminating amplification, TGS can directly evaluate a clinical specimen. The devices are handheld and affordable; approximately $1,000.
The MinION sequencer© (Oxford Nanopore Technologies, Oxford, UK) is a new-to-market palm-sized DNA sequencer that has previously been used for real-time detection of viral and bacterial pathogens in blood,82,83 urine, 84 as well as for rapid species identification in tuberculosis. 85 Reports using such a device for identification of pathogens in pneumonia sputum samples suggests TGS analysis can be performed in under 6 hours, far less than conventional culture. 86 In addition, epigenetic influences would not be lost as TGS can evaluate for the presence of methylation on the genome. 87
Several published reports have demonstrated feasibility of TGS as a tool to evaluate the DFU microbiome,88,89 highlighting its utility, but none have translated it for direct clinical application nor evaluated its sensitivity in context of conventional cultures. Additionally, tissue harvesting in the few TGS studies was performed through surgical incisions subjecting the microbiome to contamination from adjacent soft tissue.89-91 In fact, aggressive debridement may remove anaerobic species 92 and may influence microbiome analysis before it begins.
There are limitations. First, DNA from a clinical sample contains large amounts of host DNA, 79 so deciphering it from microbe DNA is required. TGS has extraordinarily long 93 outputs. While the technology is handheld and convenient for clinical application, it is reliant upon a USB cable for information transfer. The processing speed of a 3.0 USB cable is reported at 400 bp a second. More troubling is the sequencer is reported to have a high error rate. 94 Finally, the bioinformatics require advanced assistance and direct clinical interpretation is not yet feasible. While TGS is exciting, many of the limitations need to be modified before wide clinical implementation.
Conclusion
Large improvement in clinical outcomes surrounding DFU are lacking and amputation rates are rising. 3 Utilizing the pedal skin ecology to guide and dictate treatment protocols while informing clinicians of expected outcomes is rapidly evolving. Whether using conventional culture methods or emerging rapid antimicrobial identification methods, understanding the dynamic role of the microbiome are critical to address and guide future research and clinical care.
Challenges and pitfalls remain. First, distinguishing between non-infected DFU and infected DFU remains a diagnostic and therapeutic challenge. The value of consensus guidelines is limited by a lack of rigorous, consistent assessment of DFI in studies, which is compounded by a dependence on conventional culture 95 and cross-sectional design of prior studies.17,20 Given the diverse microbial taxonomy in DFU, integration of emerging technologies into clinical practice remains difficult. Therefore, it is not only crucial to discover what organisms constitute the DFU microbiome as it relates to a clinical state, but to understand the information derived from these techniques and determine how it can be translated to guide clinical care.
The existing literature confirms the complementary role of advance analysis methods to explore the complex microbiome in DFU. Further well-designed large scale studies are essential to more completely understand the data derived from these technologies and to translate the clinical and prognostic ability of the DFU microbiome, its dynamic change, and relation to healing and non-healing events.
Footnotes
Abbreviations
16S rRNA, 16S ribosomal RNA; CLSI, Clinical and Laboratory Standards Institute; DFI, diabetic foot infection;DFU, diabetic foot ulcers; DM, Diabetes mellitus; EUCAST, European Committee on Antimicrobial Susceptibility Testing; ID, infectious disease; LEA, lower extremity amputations; MALDI-TOF-MS, Matrix assisted laser desorption/ionization time-of-flight mass spectrometry; MDRO, multidrug resistant organisms; MENSA, medium-enriched for newly synthesized anti-SA antibodies; MRSA, methicillin-resistant Staphylococcus aureus; MSC, mesenchymal stromal cells; OUT, perational taxonomic units; PCR, Polymerase chain reaction; RT-PCR, real-time PCR; SA, Staphylococcus aureus; TGS, Third Generation Sequencing; WGS, Whole genome sequencing.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Grant Number 5U01DK119083 from the National Institute of Diabetes and Digestive and Kidney Disease