
Abstract
Age of onset in Huntington disease (HD) is influenced by cis-acting genetic variants, particularly the loss of interrupting codons in the HTT CAG and CCG repeats (CAG-CCG LOI variant). The CAG-CCG LOI variant is not detectable by current diagnostic assays, leading to underestimation of CAG repeat length, misdiagnosis, and inaccurate prediction of risk of symptom onset in the reduced penetrance range. In clinical trials, unidentified CAG-CCG LOI variants may affect interpretation of results, particularly for small trials. Accurate ascertainment and reporting of the CAG-CCG LOI genotype therefore has important implications for HD diagnosis, genetic counselling, and clinical trial design.
Keywords
Background
Age of onset of motor signs in Huntington's disease (HD) depends on both the length of the pathogenic CAG repeat expansion in the huntingtin gene (HTT) and the presence of onset-modifying genetic variants.1–9 Although the inherited CAG repeat length is the primary determinant of age at onset, there remains substantial variability even among patients with the same CAG repeat length. To date, rare cis-acting sequence variants which lack interrupting codons in the CAG or CCG repeat are recognized to have the largest modifying effects on onset of HD. 5 A variant characterized by simultaneous loss of interrupting codons in both CAG and CCG repeats (CAG-CCG LOI variant, Figure 1A) is the most common loss-of-interruption variant in European-ancestry HD patients, and the variant which most dramatically hastens disease onset.5,6,8
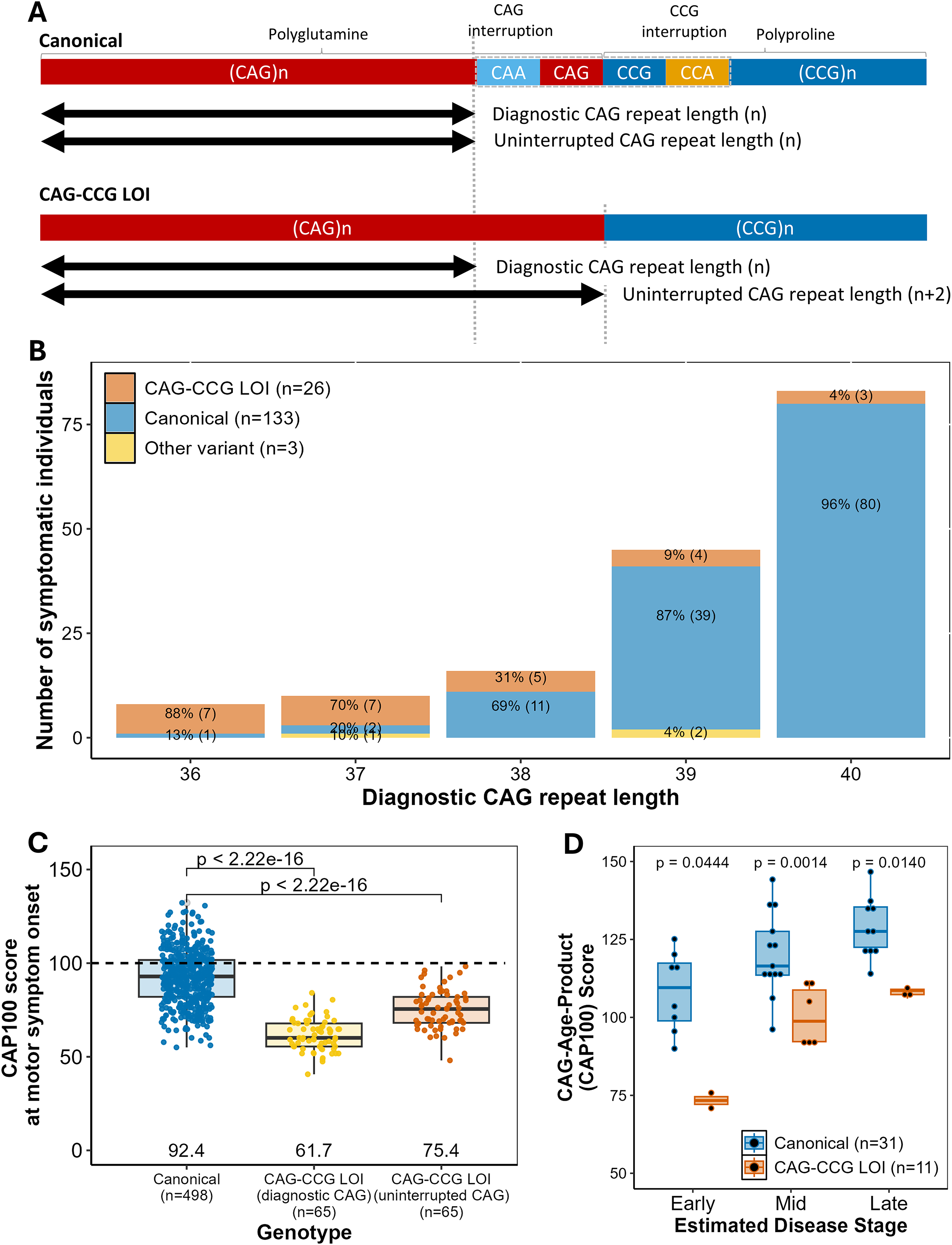
Features of the CAG-CCG LOI variant. (A) Structure of canonical and CAG-CCG LOI variant alleles. Polyglutamine-coding CAG repeat (red), polyproline-coding CCG repeat (dark blue), glutamine-coding CAA codon (light blue) and proline-coding CCA codon (yellow) are shown. Black arrows indicate diagnostic CAG repeat length (PCR fragment length), and uninterrupted CAG repeat length. (B) Frequency of CAG-CCG LOI variants in symptomatic HD. Allele frequencies for canonical (blue, n = 133), CAG-CCG LOI (orange, n = 26), and other variant alleles (yellow, n=3) for each diagnostic CAG repeat length from 36-40. CAG repeat lengths are determined by PCR fragment size, and allele structures by screening of individuals with HD motor onset from UBC HD Biobank (all CAG 36-40), and Bochum Biobank (all CAG 36-39). (C) CAP scores at motor onset for individuals with canonical or CAG-CCG LOI variant alleles. CAG-Age-Product scores at motor onset determined using the CAP100 formula for 563 individuals with known age at motor symptom onset (498 canonical, 65 CAG-CCG LOI). Mean CAP100 score was 92.4 among canonical individuals (blue). For individuals with CAG-CCG LOI variant alleles, CAP100 scores were calculated based on both diagnostic CAG repeat length (orange, mean CAP100 = 61.7), and uninterrupted CAG repeat length (yellow, mean CAP100 = 75.4). CAG repeat length and interruption sequence were determined from blood DNA. (D) CAP scores by disease stage for individuals with canonical or CAG-CCG LOI variant alleles. CAG-Age product scores at death determined using the CAP100 formula for 42 individuals with available postmortem brain tissue for analysis (31 canonical, 11 CAG-CCG LOI). CAG repeat lengths were matched between allele structures (canonical mean CAG = 42.1, CAG-CCG LOI mean CAG = 41.2), and cases were grouped by disease progression based on degree of caudate atrophy (early stage = Vonsattel Grade 1, mid stage = Vonsattel Grade 2, late stage = Vonsattel Grades 3-4).
The CAG-CCG LOI variant hastens HD onset by about 10 years relative to predicted onset based the number of uninterrupted CAG repeats.5–11 However, the CAG-CCG LOI variant is undetectable by the ACMG recommended standard PCR-fragment-length-based diagnostic assays, and causes underestimation of the number of uninterrupted CAG repeats by 2 repeat units (Figure 1A).5,8–10,12–15 This technical limitation compounds the biological onset-hastening effect, resulting in onsets on average 20 years earlier than expected based on reported CAG repeat length as underestimated from diagnostic testing (“diagnostic CAG repeat length”), with individual onsets ranging from 5–40 years earlier than predicted onset. 10 Since genetic testing for HD currently reports diagnostic rather than uninterrupted CAG repeat length, the impact of CAG-CCG LOI variants in clinical settings can greatly exceed published effect sizes based on uninterrupted CAG repeat length. The CAG-CCG LOI variant also increases disease penetrance for individuals in the reduced penetrance range, and is highly enriched among symptomatic individuals with reduced penetrance alleles (36–39 CAG repeats) even after correcting for accurate uninterrupted CAG repeat length. 8
As our understanding of the CAG-CCG LOI variant and other similar cis-acting modifiers of HD onset grows, their clinical importance has become increasingly clear, leading several groups to advocate for consideration of these variants in updates to current clinical practice.8,12,16–18 Here, we examine the implications of the CAG-CCG LOI variant on clinical diagnosis, genetic counselling, and clinical trial patient selection, to provide a resource for clinicians and genetic counsellors who may encounter patients with these variants in their practice. We focus here on the CAG-CCG LOI variant, because it is the best characterized variant and has unique implications for individuals with alleles in the reduced penetrance range, but many of the clinical concerns we outline here are also relevant to other similar variants affecting the repeat interruption sequence. Table 1 outlines characteristics of the CAG-CCG LOI variant in comparison to other variant structures.
Summary of characteristics of CAG-CCG LOI and other cis-acting repeat interruption variants.

A Findlay Black et al., Genet Med Open 2024.
B Dawson et al., Genet Med 2024.
C GEM-HD, Nat Genet, 2025.
D Findlay Black et al., Genet Med 2020.
E Reanalysis of dataset from Dawson et al., Genet Med 2024.
F Wright et al., Am J Hum Genet 2019.
G Dawson et al. Hum Genet Genomics Adv 2022. Becanovic et al. Nat Neurosci 2015.
Diagnostic implications
The most concerning implication of the CAG-CCG LOI variant is underestimation of the CAG repeat length in the genetic testing and diagnosis of HD. This occurs because PCR-based diagnostic assays consider only fragment length, but not the underlying sequence. The CAG-CCG LOI variant increases the uninterrupted CAG repeat length by 2 repeats without increasing the corresponding PCR fragment length (Figure 1A), making the change undetectable from conventional assays used for standard genetic testing of HD. To align with information currently available in clinical practice, we will discuss diagnostic CAG repeat lengths throughout this text, except where we refer specifically to uninterrupted CAG repeat length.
Among individuals with apparent intermediate allele CAG repeat lengths of 34–35 based on diagnostic testing results, those with CAG-CCG LOI variant will have true uninterrupted CAG repeat lengths of 36–37, within the pathogenic range (Table 2A). Three reported cases from independent families indicate that diagnostic CAG repeat lengths of 34–35 can cause HD when CAG-CCG LOI variants are present.8,19 It is unclear how frequent such cases may be, since these individuals are not considered at risk for HD based on current standards,14,15 and therefore may not receive HD diagnoses. Screening for these variants among individuals with HD-like symptoms whose diagnostic testing results fall in the range of 34–35 CAG repeats would assist clinicians in accurately diagnosing HD. While CAG repeat lengths of 34–35 CAG repeats are already reported by most diagnostic labs, their relevance in the context of an individual with HD-like symptoms is generally unclear. In these scenarios, presence of a CAG-CCG LOI variant may increase certainty of a suspected HD diagnosis, while the absence of such variants might prompt phenocopy investigations for an alternative cause of the patient's symptoms.
Effect of CAG repeat length underestimation of CAG-CCG LOI variants on allele classification.

The CAG-CCG LOI variant has related implications for diagnosis of HD within the currently-accepted pathogenic range for HD, particularly in the reduced penetrance range where it is unexpectedly frequent and appears to increase disease penetrance. 8 Among participants in the UBC HD Biobank with symptomatic HD and diagnostic CAG repeat lengths >40, we observe CAG-CCG LOI variant frequencies of 0.5% (6/1132), similar to reported general population allele frequencies of 0–0.18%. 20 However, CAG-CCG LOI variant frequency is highly enriched among symptomatic individuals with the shortest pathogenic CAG repeat lengths (36–39) (Figure 1B). 8
78% (14/18) of symptomatic individuals with diagnostic CAG repeat lengths of 36–37 had CAG-CCG LOI variant alleles, while canonical alleles were uncommon at these lengths in symptomatic subjects (3/18, 17%). The presence of a CAG-CCG LOI variant should therefore be strongly suspected in symptomatic individuals with diagnostic CAG repeat lengths of 36–37, and variant screening should be considered to ensure accurate reporting of uninterrupted CAG repeat length.
In contrast, only 15% (9/61) symptomatic individuals with diagnostic CAG repeat lengths of 38–39 had CAG-CCG LOI alleles. However, underestimation of the CAG repeat length of CAG-CCG LOI alleles means that individuals with diagnostic test results of 38–39 CAG in fact have uninterrupted CAG repeat lengths of 40–41, resulting in misclassification of fully-penetrant allele lengths as reduced-penetrance alleles (Table 2A).
At all pathogenic CAG repeat lengths, CAG-CCG LOI variants broadly affect HD onset and progression. Patients reach cognitive, motor, and brain imaging milestones at atypically young ages relative to their uninterrupted CAG repeat length, and may have accelerated progression of motor signs.5–11,21,22 Awareness of an individual's allele structure could enable clinicians to better anticipate and provide guidance about possible disease trajectories for a given individual.
In cases where variant allele structures are suspected, additional testing can be conducted to identify these variants. Sequencing is the most accurate method of identifying variant alleles, but where sequencing is impractical, it is possible to detect several common variant alleles through a simple alteration of standard fragment-analysis-based assays for CAG repeat length determination. 12 These assays provide the same CAG repeat length information as standard genetic testing protocols, but use an alternative reverse primer which binds across the CAG repeat interruption. This minor alteration to testing protocols generates electrophoretic traces with peak height patterns which differ between alleles with canonical and non-canonical CAG repeat interruption sequences, enabling detection of variant alleles such as CAG-CCG LOI variants.
Genetic counselling implications
At all pathogenic CAG repeat lengths, age of disease onset in HD is highly variable, making it difficult for genetic counsellors to provide accurate predictions based on CAG repeat length alone.2,3,23–26 While considering CAG-CCG LOI variant status does not enable precise prediction of disease onset on an individual basis, it is critical for diagnostic accuracy and may improve estimates for prediction of onset. CAG-CCG LOI variant status is necessary information for accurate counselling of individuals with 34–39 CAG repeats who are undergoing predictive testing for HD.
When counselling individuals with predictive testing results in the intermediate allele range, persons with 34–35 CAG repeats are currently informed that they are not at risk of developing HD. However, recent reports suggest accurate counselling in these CAG ranges cannot be provided without determining CAG-CCG LOI variant status, since these individuals may develop signs and symptoms of HD due to underestimation of CAG repeat length if a CAG-CCG LOI variant is present.8,19
When counselling individuals in the reduced penetrance range, disease outcomes are typically considered unpredictable. Recent research suggests that allele structure differences may explain much of this variability.8,22 While most asymptomatic individuals with CAG repeat lengths in the reduced penetrance range are expected to have canonical alleles, the hastening of onset in individuals with CAG-CCG LOI variants results in significant enrichment of these variants among symptomatic individuals (Figure 1B).8,20 Supplementary Table 1 further demonstrates the enrichment for CAG-CCG LOI variants among symptomatic but not asymptomatic individuals with diagnostic CAG repeat lengths from 34–39. Since CAG-CCG LOI variant status strongly influences penetrance of shorter HD-causative CAG repeat alleles, knowledge of an individual's allele structure would enable more accurate, individualized discussions of disease risk.
When counselling individuals with diagnostic CAG repeat lengths of 36–37, these individualized discussions of disease risk are particularly relevant. For example, alleles with 37 CAG repeats are only rarely associated with presentation of HD during a normal lifespan, with a previous population-based study estimating penetrance at 0.2%.8,27 However, among individuals with 37 CAG repeats who develop signs and symptoms of HD, the majority have CAG-CCG LOI variant alleles (Figure 1B). 8 An individual with a predictive testing result of 37 CAG repeats is more likely to develop signs and symptoms of HD if this is a CAG-CCG LOI variant allele than if it is a canonical allele, but current predictive testing results do not differentiate between these scenarios.
When counselling individuals with diagnostic CAG repeat lengths of 38–39, CAG-CCG LOI variant alleles still increase penetrance. Additionally, underestimation of CAG repeat length of CAG-CCG LOI variant alleles means that some individuals with fully-penetrant alleles will be counselled as though they have reduced penetrance alleles (Table 2A), potentially influencing major life choices including family-planning or careers.
Since individuals with CAG-CCG LOI variants develop motor signs of HD on average two decades earlier than expected based on incorrectly underestimated diagnostic CAG repeat length, even approximate predictions of onset will be inaccurate when variant status is not considered. 10 While screening for trans-acting modifiers of disease onset is unlikely to significantly improve onset predictions, 18 the greater impact of the CAG-CCG LOI variant means that identification of this variant is critical for providing accurate counselling in HD genetic testing, particularly for individuals in the reduced penetrance range.
When taking a family history, certain characteristics may suggest the presence of a CAG-CCG LOI variant, as outlined in Table 2B. We recommend that these characteristics prompt follow-up screening, via sequencing or using a simple alternative primer to standard fragment-analysis-based testing, 12 in cases where presence of a CAG-CCG LOI variant would affect disease classification or patient decision-making. While we have outlined several scenarios in which variant status is likely to be relevant, these are not exhaustive, and decisions about variant testing should be considered on an individual basis.
Risk factors for presence of CAG-CCG LOI variants and suggested criteria for follow-up screening.

Clinical trial implications
The mechanism causing earlier disease onset in individuals with CAG-CCG LOI variants is not yet understood, so it is unclear whether these individuals may respond differently to therapeutic intervention. Regardless of this possibility, inclusion of undetected CAG-CCG LOI variants may still affect interpretation of trial results. Individuals with CAG-CCG LOI variants may exhibit faster symptom progression, 8 and erroneous underestimation of uninterrupted CAG repeat length in these individuals would prevent proper application of selection criteria and cohort matching based on CAG repeat length.
CAG-Age-Product (CAP) score, an estimate of disease burden based on a person's age and CAG repeat length, is commonly used both in participant selection and in analysis of clinical trial results. 28 However, CAP scores are not comparable between individuals with CAG-CCG LOI and canonical alleles, due to the younger age at onset and underestimation of CAG repeat length in individuals with CAG-CCG LOI variants. At motor onset, CAP scores are significantly lower in individuals with CAG-CCG LOI variants, and this effect persists even after accounting for uninterrupted CAG repeat length (Figure 1C). Among subjects with available postmortem brain tissue for analysis, those with CAG-CCG LOI variants have lower CAP scores than canonical HD cases matched for uninterrupted CAG repeat length at early- (Vonsattel Grade 1), mid- (Vonsattel 2), and late-stage (Vonsattel 3–4) disease (Figure 1D). This indicates that a given CAP score does not represent the same disease state in individuals with and without CAG-CCG LOI variants, and therefore cannot be accurately used for comparisons involving individuals with this allele structure.
Where possible, CAG-CCG LOI variant alleles should be identified by screening or sequencing of all individuals included in a clinical trial. Variant status should be considered when balancing treatment groups, uninterrupted CAG repeat length should be used for all analyses, and individuals with variant alleles should be excluded from analyses involving CAP score, until variant status is accounted for in an updated CAP score measure. If identification of CAG-CCG LOI variants is not feasible in a therapeutic trial, the trial should be designed to reduce the impact of unidentified variant alleles on trial interpretation. Given the rarity of CAG-CCG LOI variants associated with CAG repeat lengths above 39, exclusion of individuals with reduced penetrance CAG repeat lengths (<40) from clinical trials is likely sufficient to prevent the presence of unidentified variant alleles from affecting interpretation of results. This is of particular importance in trials including small numbers of individuals, such as gene therapy trials.
Key messages
Diagnosis and genetic counselling
Current genetic testing does not detect or consider the CAG-CCG LOI variant or other cis-acting genetic variants.
Allele structure, not just CAG repeat length, is necessary for accurate allele classification and diagnosis of HD.
CAG-CCG LOI variant status affects disease penetrance and disease course, and should be considered when providing genetic counselling, particularly for individuals with 34–39 CAG repeats.
Clinical trials
In clinical trials, particularly those with small numbers of participants, individuals with sequence variants should be accounted for in balancing and analysis. If direct identification is not possible, excluding individuals with CAG <40 reduces the likelihood that CAG-CCG LOI variants may affect interpretation of trial results.
CAP scores are inaccurate for comparing between individuals with canonical and CAG-CCG LOI variant allele structures. An updated CAP score measure that accounts for variant status is needed.
All clinical practice
While the CAG-CCG LOI variant is the best characterized and most common sequence variant, many of the same concerns apply to other cis-acting variants, as outlined in Table 1.
Sequencing is the most accurate method of identifying variant alleles, but where sequencing is impractical, it is possible to detect several common variant alleles through a simple alteration of standard fragment-analysis-based assays for CAG repeat length determination. 12
Supplemental Material
sj-docx-1-hun-10.1177_18796397261443135 - Supplemental material for Clinical implications of loss of interruption variants for diagnosis, genetic counselling, and clinical trials in Huntington's disease
Supplemental material, sj-docx-1-hun-10.1177_18796397261443135 for Clinical implications of loss of interruption variants for diagnosis, genetic counselling, and clinical trials in Huntington's disease by Hailey Findlay Black, Jessica Levesley, Chris Kay, Stephanie Bortnick, Kyla Javier and Michael R Hayden in Journal of Huntington's Disease
Footnotes
Acknowledgements
The authors thank the HD patients and families who have participated in research through sample and data contributions. Brain tissues examined in this study were received from the University of British Columbia HD Biobank, the University of Washington Biorepository and Integrated Neuropathology (BRaIN) Laboratory, and the Centre for Brain Research at the University of Auckland. Blood DNA samples were received from the University of British Columbia HD Biobank and the Ruhr University Bochum.
Ethical considerations
Participant samples and clinical data were collected with informed consent and ethical approval from the UBC Children's and Women's Health Centre Research Ethics Board (H06-70467, H05-70532), the Ruhr University Bochum Medical Faculty Ethics Committee (18-6563-BR), and the New Zealand Health and Disability Ethics Committee (HDEC 20/NTA/166).
Generative AI was not used for any aspects of the work.
Consent to participate
Participant samples and clinical data were received in anonymized format from the University of British Columbia HD Biobank, the University of Washington Biorepository and Integrated Neuropathology (BRaIN) Laboratory, the Centre for Brain Research at the University of Auckland, or the Ruhr University Bochum, and were collected with written informed consent by their respective biobanks.
Consent for publication
Not applicable.
Author contributions
Findlay Black, H analysed data, generated figures, and wrote the manuscript. Bortnick, S and Javier, K curated clinical data and assisted with manuscript editing. Kay, C, Levesley, J, and Hayden, MR assisted with manuscript editing, figure design, and suggestions for analysis.
Funding
This work was supported by a Canadian Institutes of Health Research Foundation grant awarded to M.R.H. (GR005171), the James Family, and the BC Children's Hospital Research Institute.
Declaration of conflicting interest
Michael R. Hayden is the CEO of Prilenia Therapeutics, a private company, and serves on the public boards of Ionis Pharmaceuticals, Oxford Biomedica, AbCellera and 89bio. All other authors declare no conflicts of interest.
Data availability
Data will be made available on request (contact Dr Michael R Hayden, mrh@cmmt.ubc.ca).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.