
Abstract
Background
Gene silencing is widely recognized as a promising therapeutic approach for dominant monogenic disorders. Current silencing strategies, many of which are transient, utilize RNA interference. Gene silencing may also be achieved through directed epigenetic editing using a CRISPR/dCas9 effector fused to DNA methyltransferase 3A (dCas9-DNMT3A). We used this system to direct DNA methylation to HTT, the causal gene underlying the autosomal dominant neurodegenerative disorder Huntington's disease, to assess the translational potential of this strategy for treating a genetic neurological disease.
Objective
To characterize the regulatory effect of targeted dCas9-DNMT3A-mediated DNA methylation at HTT.
Methods
We exploited DNA methylation profiles of high and low HTT-expressing tissues and targeted hypomethylated regions of HTT associated with high levels of HTT expression.
Results
De novo DNA methylation of loci within defined upstream, promoter, intragenic and downstream regions of HTT resulted in robust, acute silencing of HTT. The best long-term silencing of HTT, which persisted up to 30 days, was observed when targeted DNA methylation was directed to the 5’UTR and promoter regions of HTT.
Conclusions
HTT gene silencing may be achieved via targeted de novo DNA methylation within hypomethylated regulatory regions at the HTT locus. DNA methylation editing may be an attractive therapeutic approach for Huntington disease due to its potential for long-term silencing and reversibility.
Introduction
Dominant monogenic disorders, although defined genetically, involve a cascade of complex cellular pathologies whose relative importance is difficult to ascertain. As a result, therapies designed to correct these dysregulated processes often have negligible impact on mutation-associated phenotypes. Alternatively, silencing of pathogenic genes is an attractive therapeutic strategy for many of these conditions because this approach directly targets the genetic origin of downstream pathogenesis. One example is the neurodegenerative disorder Huntington's disease (HD), which is characterized by a triad of cognitive, motor and behavioral disturbances. Principally, these abnormalities are caused by highly penetrant mutations in the CAG triplet repeat tract in exon 1 of the huntingtin (HTT) gene, which encodes a mutant form of the huntingtin protein (HTT). 1 Studies using genetically modified mouse models of HD have shown that lowering mutant HTT, even at advanced disease stages, ameliorates HD-like phenotypes.2,3 Hence, therapeutic approaches focused on reducing mutant HTT abundance are of great interest.
Strategies designed to lower mutant HTT, either selectively or together with wild-type HTT, include RNA interference (RNAi) and antisense oligonucleotides (ASOs). 4 Despite promising preclinical results, recent evaluations of these interventions in clinical trials suggest limited therapeutic benefit and indicate concomitant toxicities. 5 These approaches, which are transient, may also necessitate frequent or continuous administration of therapeutic nucleic acids, which is an undesirable requirement given the chronic nature and already significant burden of HD. The advent of CRISPR/Cas9 genome editing technology has created enormous opportunities for developing alternative and highly durable therapeutic gene silencing approaches. 6 In the context of HD, CRISPR/Cas9-based therapeutic strategies involve permanent genome editing of the mutation containing-region of HTT.7–10 While this approach has the potential to permanently lower HTT, any accompanying side effects resulting from off-target editing, including at other CAG repeat-containing genes, would also be irreversible.
In contrast, gene silencing induced by epigenetic modifications, such as DNA methylation, may induce significant regulatory effects that are both durable and reversible, as suggested by the utility of this approach for cancer treatment. 11 The CRISPR/Cas9 system has been adapted for DNA methylation editing and enables deposition of targeted, genome-wide DNA methylation using catalytically inactive Cas9 tethered to a de novo DNA methyltransferase enzyme (dCas9-DNMT3A). 12 The regulatory effects of CRISPR/Cas9-mediated DNA methylation editing have been evaluated at genes implicated in several neurological disorders, including autism spectrum disorder, 13 Alzheimer's disease, 14 and Parkinson's disease. 15 In these studies, targeted DNA methylation editing within regulatory regions and regions in which DNA methylation patterns are altered by disease state demonstrated significantly reduced expression of the disease-linked genes Mecp2, APP, and SNCA, highlighting both the utility and therapeutic relevance of this intervention.13–15 While studies using CRISPR/Cas9-based epigenetic silencing methods have primarily targeted gene promoter regions, rational approaches for identifying specific loci to induce optimal DNA methylation-based epigenetic silencing remain to be established.12,16 Systematic identification of additional sites for de novo DNA methylation would be extremely beneficial for single nucleotide polymorphism (SNP)-based allele-specific strategies for epigenetic silencing, which may permit selective mutant HTT lowering. To address this, we reasoned that such putative sites could be identified using DNA methylation profiles of tissues associated with high or low expression of a particular gene. Focusing on HTT, we utilized this strategy to identify multiple loci within the upstream, promoter, intragenic and downstream regions of HTT and targeted them using pools of guide RNAs (sgRNAs) to induce site-specific DNA methylation.
Methods
sgRNA design and generation
sgRNAs were designed in close proximity to CTCF binding sites and CpG islands. The CTCFBSDB 2.0 database 17 was used to predict CTCF binding sites within the HTT promoter defined by Coles et al. 18 and Eukaryotic Promoter Database 19 as the region within −499 bp to +1 with respect to the TSS. Predicted CpG islands within the intragenic region were annotated based on ≥50% GC content, lengths exceeding 200 bp, and observed/expected CpG dinucleotide ratios >0.6 (UCSC Genome Browser). sgRNAs 20 nucleotides in length were designed manually using the HTT gene sequence from UCSC Genome Browser (Human GRCh38/hg38) and a modified procedure developed by Vojta et al. 12 Target sequences were selected within 10–100 bp upstream of sites targeted for DNA methylation, and pooled sgRNAs were used to achieve a synergistic effect. 12 sgRNAs with predicted off-target binding sites with at least 1 mismatch and closer to PAM sequences were selected, and the core length was 9–12 nucleotides. Off-targets were checked using GT-Scan and CRISPOR.20,21 sgRNA sequences are shown in Table 1, and computationally-predicted sgRNA efficiencies and off-target analyses are shown in Supplemental Table 2.
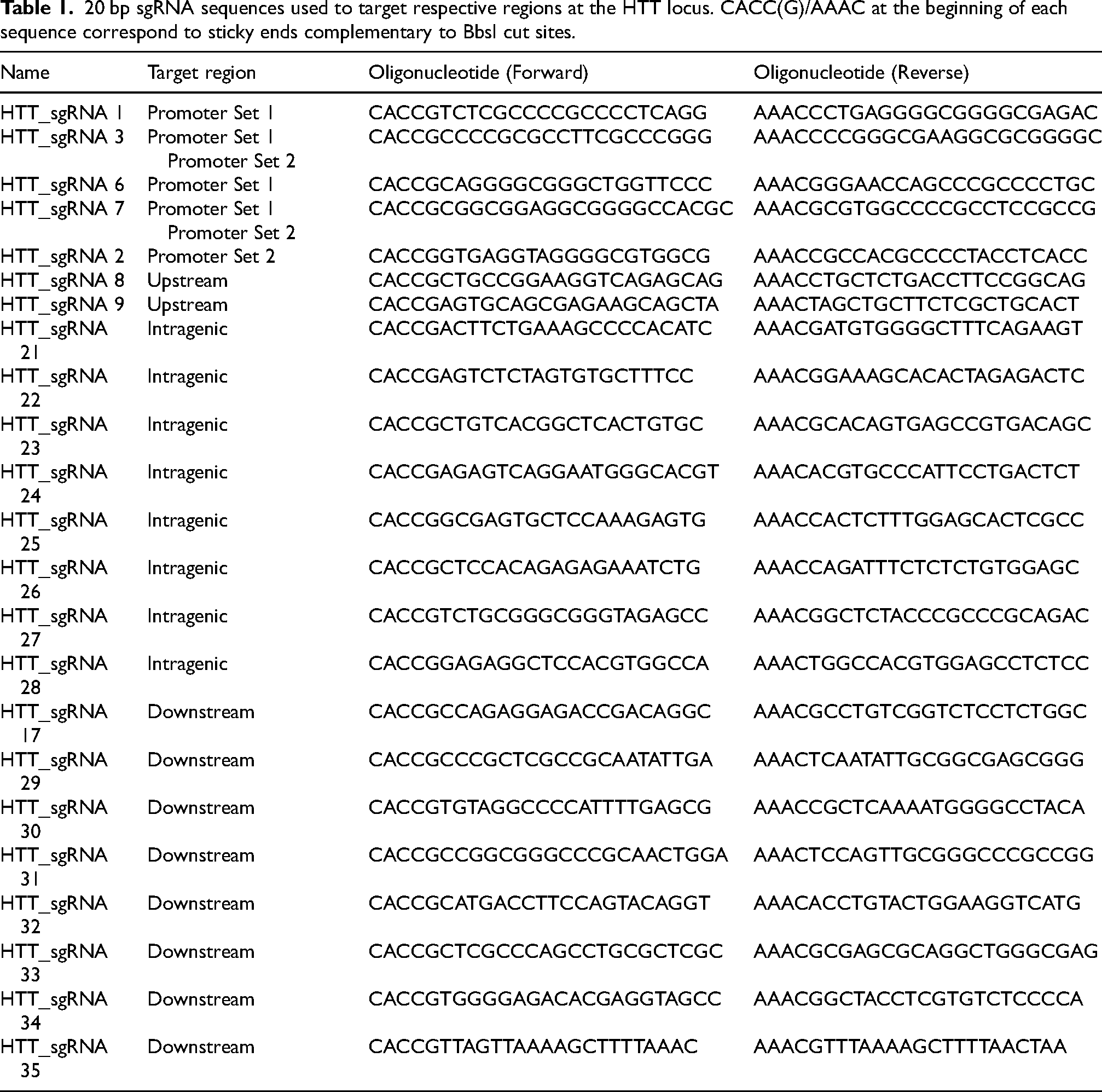
20 bp sgRNA sequences used to target respective regions at the HTT locus. CACC(G)/AAAC at the beginning of each sequence correspond to sticky ends complementary to BbsI cut sites.
pdCas9-DNMT3A-PuroR_v2 was a gift from Vlatka Zoldoš (Addgene plasmid #74407; http://n2t.net/addgene:74407; RRID:Addgene_74407). 12 The GFP sgRNA plasmid was a gift from George Church (Addgene plasmid # 41819). The plasmid was purified using QIAGEN® Plasmid Maxi kit (QIAGEN, Cat No.: 12162) according to the manufacturer's instructions. sgRNA oligonucleotides were synthesized by Integrated DNA Technologies, Inc. 1 µL each of forward and reverse sgRNA oligonucleotides (100 µM) were annealed using 0.5 µL of T4 PNK (Promega), 2 µL T4 10X ligation buffer and 10 mM ATP in 10 µL total volume (37°C 30 min, 95°C 5 min, ramping 0.2°C/s to 25°C). For each pair of annealed sgRNA oligonucleotides, 1 µL of 1/100 diluted annealing reaction was ligated with 25 ng of the purified BbsI-digested vector plasmid using 1 µL T4 DNA ligase, 0.6 µL T4 10X ligation buffer, and 10 mM ATP in 6 µL total volume and incubated (1 h, 37°C). Plasmid constructs were transformed into OneShot® TOP10 competent E. coli (ThermoFisher Scientific) following the manufacturer's protocol. Cloned sgRNA sequences were verified using Sanger sequencing and correct clones were isolated using the QIAprep Spin Maxiprep kit (QIAGEN, Cat No.: 27106).
Tissue culture
HEK293T cells were obtained from ATCC (CRL-3216). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum, 1% 100 U/mL penicillin and 10 µg/mL streptomycin and passaged with 1X trypsin/EDTA solution up to passage 30. Cells were incubated at 37°C in a humidified chamber with 5% CO2.
For transfection, cells were seeded in 6-well plates at a density of 106 cells/well. Using a total of 2 µg plasmid DNA per well, transfection was performed 1-day post-seeding using jetPRIME reagent (Polyplus) according to the manufacturer's instructions. There were 12 biological replicates for each set of transfections. 1.5 µg/mL of puromycin-containing media was added to each well 1 day post-transfection and puromycin selection lasted for 2 days (the time required for complete cell death of untransfected cells). Thereafter, transfected cells and mock-transfected cells were maintained in normal culture media until harvesting at day 7 or day 30 post-transfection. Harvested cultures were centrifuged (3000 rpm, 3 min, 4°C) and washed twice with 1X PBS (Gibco). One half of each cell pellet was used for RNA extraction and the remainder was processed for pyrosequencing.
RNA extraction for real-time quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen). For real-time quantitative RT-PCR, harvested RNA was used to perform first-strand cDNA synthesis using the SuperScript First Strand Synthesis System (Invitrogen). All probes were run in triplicate with SYBR Select Universal Master Mix (Invitrogen) on a StepOnePlus instrument (ABI) to obtain Ct values. Relative gene expression levels were analyzed using the Comparative CT Method (ΔΔCT Method). The primers used are as follows: beta-actin (F): GGCATGGGTCAGAAGGATTC, beta-actin (R): CACACGCAGCTCATTGTAGAAG, HTT (F): GCACCGACCAAAGAAAGAAC, HTT (R): TCATCACTGCACAGCAGAAA.
Pyrosequencing analysis of regional DNA methylation
To characterize patterns of regional DNA methylation, pyrosequencing was performed on regions of interest at the HTT locus in 48 samples. A total of 32 CpG sites were assayed in the region (chr4:3,071,446–3,239,627; hg38). Genomic DNA (gDNA) was extracted from transfected cells using the DNeasy Blood & Tissue kit (QIAGEN, Cat No.: 69504). gDNA was eluted using nuclease-free water and concentrations were adjusted to 25 ng/uL. Pyrosequencing was performed using biotinylated primers to amplify bisulfite-treated DNA (Supplemental Table 1). Biotinylated PCR products were immobilized on streptavidin-coated Sepharose beads (GE Healthcare, Orsay, France). Pyrosequencing was performed with the PyroMark Q96 MGMT kit (Qiagen, Courtaboeuf, France) on a PSQTM96 MA system (Biotage, Uppsala, Sweden).
Statistics
Statistical significance was evaluated by 1-way ANOVA with Bonferroni's multiple comparisons test (Figure 1B-G) or unpaired t-test (Figure 2C, D) using GraphPad Prism V6 and V10.4.2, respectively. Outliers were omitted based on the D’Agostino-Pearson omnibus normality test. Not significant (n/s), *p < 0.05, **p < 0.01, ***p < 0.001. All data are reported as mean ± SEM unless otherwise indicated.
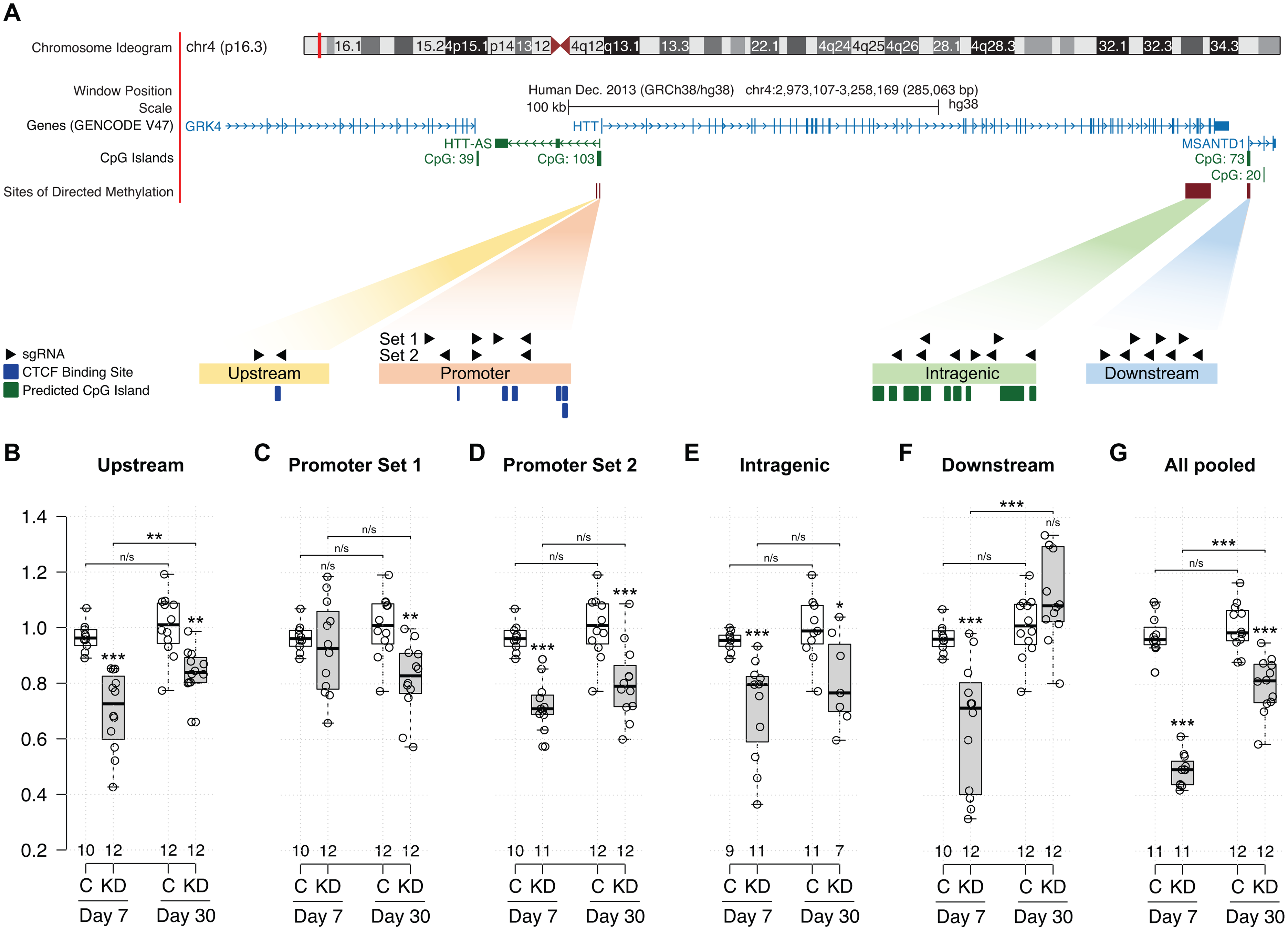
Targeted de novo DNA methylation of HTT results in sustained HTT knockdown. (A) Schematic of the HTT gene and surrounding regions targeted with dCas9-DNMT3A. For each site to which DNA methylation was directed, sgRNA positions and orientations are indicated by black arrows. Positions of predicted CpG islands and CTCF binding sites are shown relative to sgRNAs. Adapted from UCSC Genome Browser. (B-G) Levels of HTT mRNA at day 7 and day 30 post-transient transfection with dCas9-DNMT3A and sgRNAs targeting the (B) upstream, (C-D) promoter, (E) intragenic, (F) downstream, or (G) all regions in the vicinity of the HTT gene. Mean ± SEM; N = 7–12 replicates/condition/day. *p < 0.05; **p < 0.01; ***p < 0.001, determined by 1-way ANOVA with Bonferroni post-hoc analysis.
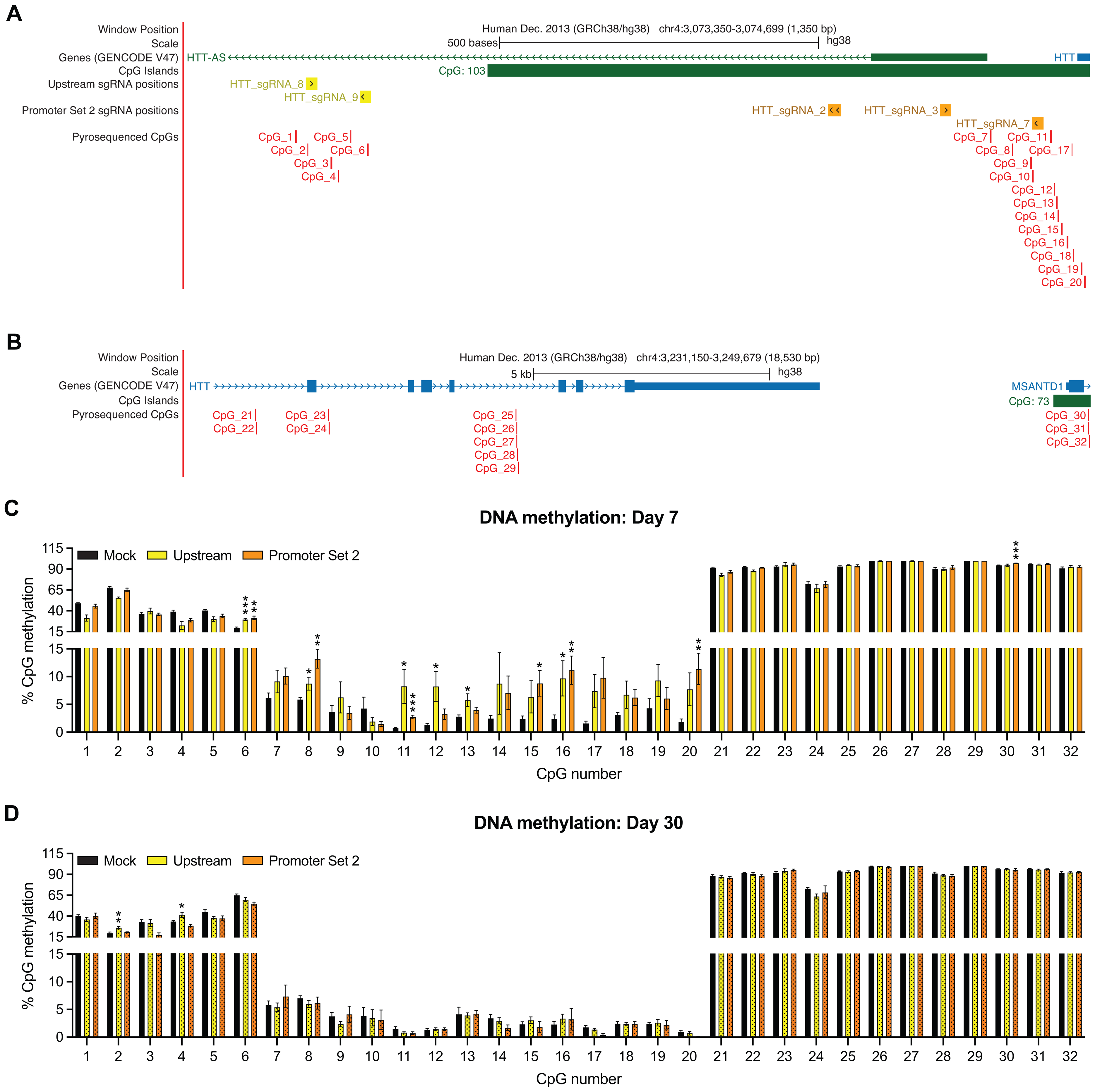
dCas9-DNMT3A induces specific, robust DNA methylation at sgRNA-targeted loci within HTT. (A-B) The positions of upstream and promoter set 2 sgRNA target sequences are shown relative to pyrosequenced CpGs and HTT. Adapted from UCSC Genome Browser. (C-D) %CpG DNA methylation levels in cells treated with dCas9-DNMT3A and (C) upstream or (D) promoter set 2 sgRNAs at day 7 and day 30 post-transfection relative to mock-transfected (unmethylated) controls. Mean ± SEM; N = 3–6 replicates/condition/day. *p < 0.05; **p < 0.01; ***p < 0.001, determined by unpaired t-test.
Results
Identification of specific regions for targeted de novo methylation of HTT
Based on the differential DNA methylation profiles of high HTT-expressing cortical tissue versus low HTT-expressing liver tissue characterized by De Souza et al., 22 we identified hypomethylated regions of HTT that correspond with high levels of HTT expression. We hypothesized that de novo DNA methylation of these hypomethylated regions using dCas9-DNMT3A 12 would epigenetically silence HTT. Four hypomethylated regions at the HTT locus were chosen as sites for targeted DNA methylation: 1) A region 1.25 kb upstream of the transcription start site (TSS); 2) the HTT promoter region defined by Coles et al. 499 bp upstream of the TSS 18 ; 3) the intragenic region between exon 60 and exon 66 of HTT, and 4) the 5’ region of the MSANTD1 gene downstream from HTT (Figure 1A, Supplemental Figure 1).
When designing sgRNAs to target these regions, we also considered locations of characterized and predicted CTCF binding sites and CpG islands, and the directionality of CpG islands within these regions. The DNA methylation statuses of CTCF binding sites and CpG islands have important effects on gene expression, 23 and targeted CRISPR/Cas9-mediated DNA methylation has been shown to disrupt CTCF binding and alter transcriptional activity.24,25 The upstream region we identified contains a CTCF binding site within the HTT promoter that is differentially methylated between cortex and liver tissues 22 (Figure 1A, Supplemental Figure 1B). DNA methylation levels are lower (18.7–25.2%) at this site in cortex, and CTCF occupancy and HTT expression are significantly higher in cortex than in liver (48.6–91.4%), demonstrating the direct impact of DNA methylation on HTT transcription and suggesting targeted DNA methylation at this site may alter HTT expression. We also identified multiple additional predicted CTCF binding sites within the HTT promoter region (Figure 1A, Supplemental Figure 1C, D). Within the intragenic region, we identified 9 CpG islands (cg19319433-cg02197303 towards the HTT 3’UTR; Figure 1A, Supplemental Figure 1E) that are hypomethylated in cortex compared to liver. 22 The downstream region also contains an 820 bp CpG island (CpG 73; UCSC Genome Browser) located within the MSANTD1 gene (Supplemental Figure 1F). Based on De Souza et al., 22 this region was included in our analysis because most hypomethylated CpGs within cortical HTT (as compared to liver HTT) are located towards the HTT 3’UTR. Because pools of sgRNAs have been shown to improve silencing efficacy, we designed multiple sgRNAs to target each region (Table 1).
Targeted de novo methylation of HTT results in sustained HTT knockdown
We assessed the regulatory impact of DNA methylation editing at HTT using human embryonic kidney (HEK293 T) cells. Previous work evaluating the impact of DNA methylation editing on gene expression in dividing peripheral cells and post-mitotic primary neurons has shown consistent regulatory effects in both model systems, validating our investigation of DNA methylation editing effects at HTT using this cellular model. 14 We transfected HEK293 T cells with pools of plasmids co-expressing dCas9-DNMT3A and sgRNAs to target selected regions within the HTT gene for de novo DNA methylation (Table 1). Mock-transfected cells were used as negative controls, and puromycin treatment was used to select for transfected cells. HTT mRNA expression was evaluated 7 days post-transfection, which is a time point expected to coincide with the peak of DNA methylation-induced downregulation. 12 To evaluate the durability of our approach, HTT expression was also assessed at 30 days post-transfection. Previous studies have only monitored the stability of CRISPR/Cas9-based DNA methylation editing up to post-transfection day 20, but indicate modified DNA methylation persists through serial passaging.16,25
This targeted approach led to downregulation of HTT expression, with region-specific differences in both efficacy and stability (Figure 1B-F). There was no effect of dCas9-DNMT3A alone or dCas9-DNMT3A in combination with a non-targeting control sgRNA on HTT expression (Supplemental Figure 2), demonstrating the HTT downregulation we measured was the result of HTT targeting. Strikingly, treatment with dCas9-DNMT3A and upstream-, promoter set 2-, intragenic-, and downstream-targeted sgRNAs significantly reduced HTT mRNA (27–35%) at day 7 post-transfection compared to mock-transfected controls, despite the lack of characterized regulatory elements in several of these regions. 26
Curiously, although no significant decrease in HTT mRNA was observed using promoter set 1 sgRNAs at day 7, there was a significant (∼19%) reduction in HTT mRNA levels measured at post-transfection day 30 (Figure 1C). Sustained silencing of HTT at day 30 was also observed with promoter set 2 sgRNAs (∼20% decrease compared to the mock-treated control) (Figure 1D). In contrast, HTT lowering that resulted from treatment with sgRNAs targeted to the downstream region was not long-lasting, as HTT mRNA levels in the treated group did not differ significantly from the control at day 30 post-transfection (Figure 1F). Pooling the sgRNAs for all targeted regions improved silencing efficacy relative to the pools of sgRNAs targeting individual regions, and led to the greatest decrease in HTT mRNA abundance (∼50% at day 7 compared to the mock-treated control) (Figure 1G). This reduction in HTT mRNA was also relatively stable: statistically-significant HTT knockdown (∼20%) was measured at 30 days post-transfection (Figure 1G).
Pyrosequencing analyses of de novo HTT methylation
To confirm these transcriptional changes were the result of DNA methylation editing, pyrosequencing of genomic DNA was performed on samples transfected with pooled plasmids co-expressing dCas9-DNMT3A and pools of sgRNAs targeting the upstream and promoter set 2 regions. We hypothesized that DNA methylation at CpGs proximal to the target sites of these sgRNAs was correlated with the HTT lowering we measured, as only treatment with upstream and promoter set 2 sgRNA pools induced significant HTT lowering at both time points (Figure 2A). We also sequenced a cluster of CpGs within the HTT 3’-end and the MSANTD1 5’ CpG island as a control (Figure 2B), and compared CpG DNA methylation levels in these samples to mock-transfected controls at day 7 and day 30.
At day 7, treatment with upstream sgRNAs increased DNA methylation largely downstream from predicted sgRNA binding sites (at CpGs 6, 8, 11–13 and 16) (Figure 2A, C). A similar pattern was observed for the promoter set 2 sgRNAs: significant increases in DNA methylation were measured at CpGs 6, 8, 11, 15, 16, 20 and 30, most of which are downstream from the most distally-positioned sgRNA in the pool (sgRNA 7) (Figure 2A, C). DNA methylation was largely unchanged at CpGs in closest proximity to sgRNA target sequences: for upstream sgRNAs, CpG 2 overlaps with the target sequence of sgRNA 8; for promoter set 2 sgRNAs, CpGs 9 and 10 are, respectively, immediately adjacent to and overlapping with the sgRNA 7 target sequence (Figure 2A). A notable exception to this trend is CpG 6, which directly overlaps with sgRNA 9 and was significantly hypermethylated following treatment with upstream sgRNAs. The increased DNA methylation measured at day 7 was no longer detectable at the same CpGs at day 30; however, treatment with upstream sgRNAs was associated with increased DNA methylation at CpGs 2 and 4 at this time point (Figure 2D).
Discussion
This is the first study examining the direct impact of DNA methylation editing on HTT gene expression. Our strategy utilized CRISPR/Cas9-mediated DNA methylation editing to hypermethylate HTT, which resulted in robust, sustained silencing of HTT.
sgRNAs were designed to direct dCas9-DNMT3A to upstream, promoter, intragenic, and downstream regions relative to HTT. These loci are hypomethylated in tissues with high HTT expression and contain CTCF transcription factor binding sites and CpG islands, which are DNA methylation-sensitive regulatory elements. Pools of sgRNAs targeting each region successfully downregulated HTT expression 27–35% at 7 days post-transfection, demonstrating that employing endogenous DNA methylation data to identify hypomethylated target regions for DNA methylation editing may be a useful general approach for lowering gene expression that can be applied to other gene or disease contexts. While greater levels of knockdown may be required to achieve clinically meaningful phenotypic improvements in the context of other disorders, the knockdown achieved here is a promising starting point for further optimization that, importantly, is aligned with clinical therapeutic ranges for HTT lowering. 27
We also assessed the persistence of HTT lowering and found that only treatment with upstream and promoter-targeting sgRNAs reduced HTT expression (19–20%) 30 days following dCas9-DNMT3A treatment. Pooling all sgRNAs also significantly lowered HTT at both time points; at day 7, the ∼50% lowering we measured appeared to be an additive effect of all sgRNAs, while the ∼20% lowering measured at day 30 was likely driven by the upstream and promoter sgRNAs. In all, our results indicate that while targeting dCas9-DNMT3A to regions within and proximal to HTT resulted in relatively comparable levels of HTT silencing, there were inherent differences in the persistence of this regulatory effect at different genomic loci. Moreover, targeting dCas9-DNMT3A to the upstream and promoter regions of HTT resulted in robust, stable HTT mRNA reduction for at least 30 days, highlighting the potential of this strategy for long-term HTT lowering.
Pyrosequencing was used to correlate changes in HTT expression with DNA methylation editing at sgRNA-targeted upstream and promoter set 2 regions. At day 7 in both regions, the most robust increases in DNA methylation were consistently measured at CpGs downstream from sgRNA target sequences. Interestingly, this pattern occurred regardless of sgRNA directionality, as both pools contained a mixture of sequences targeted to + and - strands. Minimal DNA methylation changes were detected at CpGs overlapping with or closely adjacent to sgRNA target sites except at CpG 6, which overlaps with the sgRNA 9 target site (Figure 2A). Of the pyrosequenced CpGs in closest proximity to the upstream sgRNA target loci (CpGs 1–6), CpG 6 was the least methylated in the mock-transfected control samples, with 19.4% CpG methylation measured at CpG 6 compared to 36.3–67.9% CpG methylation at CpGs 1–5 (Figure 2C). Consistent increases in DNA methylation were also measured at CpGs 11–20, which were similarly lowly-methylated (0.8–4.3%) in the mock-transfected control samples, supporting the hypothesis that targeted DNA methylation editing at hypomethylated regions of HTT is an effective HTT lowering strategy.
Despite targeting dCas9-DNMT3A to hypomethylated regions of HTT reported by De Souza et al., 22 pyrosequencing revealed that CpGs 21–32 within the intragenic and downstream regions were nearly 100% methylated in mock-transfected control samples (Figure 2C, D). This finding emphasizes the importance of careful model system selection, as endogenous in vivo HTT DNA methylation patterns were not fully replicated in vitro in HEK293 T cells. Nevertheless, our results demonstrate that targeting dCas9-DNMT3A to hypomethylated HTT DNA sequences using sgRNAs upstream from target CpGs effectively edits DNA methylation and downregulates HTT.
While the HTT lowering induced by upstream and promoter set 2 sgRNA treatment at day 7 was also detectable at day 30, edited DNA methylation was not (Figure 2C, D). Increased DNA methylation was measured on day 30 at CpGs 2 and 4 in the upstream sgRNA-treated group (Figure 2D); however, these increases were not present at day 7 (Figure 2C), indicating that consistent edited DNA methylation was not correlated with the reduced HTT expression we observed. In this context, regulatory mechanisms aside from DNA methylation editing may also contribute to HTT lowering. Targeting of dCas9 to promoter loci has been shown to reduce gene expression, demonstrating that dCas9-associated steric hindrance may be sufficient to modulate transcription in the absence of additional effectors. 28 dCas9-DNMT3A targeting may also recruit other epigenetic regulators that alter gene expression through DNMT3A-independent mechanisms—for example, MeCP2, which directly interacts with and inhibits DNMT3A, 29 is a well-characterized transcriptional repressor that binds methylated DNA and stimulates gene silencing. 30 Alternatively, targeted DNA methylation editing may catalyze downstream regulatory changes that may be more durable than the edited DNA methylation itself. In part, the stability of epigenetic silencing is dependent on factors that govern the rate of demethylation that are, at present, poorly understood; fortunately, advancing methodologies are becoming more capable of monitoring such silencing processes in vitro. 6
Although the sgRNAs used in this study target both wild-type and mutant HTT alleles, future work should explore SNP-based sgRNA design strategies that could enable selective targeting of the mutant allele. Such allele-specific approaches would represent an important advancement toward epigenetic silencing therapies for HD, potentially allowing for preservation of wild-type HTT function while specifically reducing mutant protein levels. Additionally, future studies should investigate the relationship between HTT knockdown efficiency and CAG tract somatic instability, as recent evidence suggests that HTT lowering can reduce somatic expansion of HTT's CAG repeats. 28 This finding raises the intriguing possibility that epigenetic HTT silencing may provide dual therapeutic benefits: direct reduction of mutant HTT expression and stabilization of the underlying CAG repeat instability that drives disease progression. Systematic evaluation of this approach in disease-relevant neuronal models, particularly HD patient-derived neurons where native chromatin architecture and DNA methylation patterns may influence target accessibility and silencing efficacy, along with assessment of the correlation between treatment efficacy and repeat stability, will be critical for advancing this therapeutic strategy toward clinical applications.
Using targeted de novo DNA methylation as a tool for manipulating the genome may improve our understanding of regulatory processes and DNA methylation dynamics, and help better inform the efficacy of epigenetic silencing-based therapeutic interventions. Besides improving epigenetic silencing efficiency and durability, CRISPR/Cas9-based DNA methylation editing strategies must also address valid concerns about potential off-target effects before they can become viable therapeutic candidates. While off-target editing can be minimized by careful in silico screening of sgRNA sequences, preclinical experimental assessment of off-target editing is a critical component of sgRNA selection that must be undertaken before clinical evaluation proceeds.
Supplemental Material
sj-docx-1-hun-10.1177_18796397251415368 - Supplemental material for Silencing of human HTT by targeted CRISPR/dCas9-mediated epigenetic editing
Supplemental material, sj-docx-1-hun-10.1177_18796397251415368 for Silencing of human HTT by targeted CRISPR/dCas9-mediated epigenetic editing by Yi Lin Tay, Sarah B Thomson, Silvia Hnatova, Sherwin Ng, Si Rui Teo, Ryan McCallum, Bernice Sim, Letizia Tarantini, Fei Li Tai, Valentina Bollati, Marie Loh, Michael R Hayden, Blair R Leavitt and Mahmoud A Pouladi in Journal of Huntington's Disease
Supplemental Material
sj-xlsx-2-hun-10.1177_18796397251415368 - Supplemental material for Silencing of human HTT by targeted CRISPR/dCas9-mediated epigenetic editing
Supplemental material, sj-xlsx-2-hun-10.1177_18796397251415368 for Silencing of human HTT by targeted CRISPR/dCas9-mediated epigenetic editing by Yi Lin Tay, Sarah B Thomson, Silvia Hnatova, Sherwin Ng, Si Rui Teo, Ryan McCallum, Bernice Sim, Letizia Tarantini, Fei Li Tai, Valentina Bollati, Marie Loh, Michael R Hayden, Blair R Leavitt and Mahmoud A Pouladi in Journal of Huntington's Disease
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
ORCID iDs
Ethical considerations
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MAP was supported by grants from the Agency for Science, Technology and Research (Singapore), Canadian Institutes of Health Research (CIHR No. ENG-191555), and the Huntington's Disease Foundation. MAP is the recipient of a BC Children's Health Research Institute Investigator Grant Award (IGAP) and a Scholar Award from the Michael Smith Health Research BC. SH was supported by the Singapore International Pre-Graduate Award from the Agency for Science, Technology and Research.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.