
Abstract
Poly(ADP-ribosyl)ation (PARylation), a crucial post-translational modification, is catalyzed by ADP-ribosyltransferases (ARTs) and has significant implications in various cellular processes, including DNA damage response, cell signaling, and immune processes. Aberrant PAR signaling is implicated in numerous neurodegenerative diseases, including Alzheimer, Parkinson, amyotrophic lateral sclerosis, and cerebellar ataxia, where increased PAR levels and PARP1 activity are commonly observed. However, Huntington disease exhibits a unique characteristic: reduced PAR levels and impaired PARP1 activity even in prodromal phase. This finding challenges the prevailing understanding of PAR's role in neurodegeneration and suggests that dysregulation of PAR signaling, whether through overactivation or suppression, can lead to neuronal dysfunction. Herein, we discuss how this balance may impact neurodegenerative diseases, and possible connections between PAR signaling and emerging modifiers of disease onset identified by HD genome-wide association studies (GWAS).
Poly(ADP-ribosyl)ation (PARylation) is a complex post-translational modification with critical roles in DNA damage response, cell signaling, and immune processes. It is a reversible post-translational modification in which ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) are transferred onto target proteins. This reaction is primarily catalyzed by enzymes known as poly(ADP-ribose) polymerases (PARPs), with PARP1 being the most extensively studied member of the family. The modification involves the addition of ADP-ribose units, either singly (MAR) or as polymers (PAR), to target molecules, with the structural diversity of PAR chains—varying in length and branching—impacting cellular physiology. 1 Emerging evidence underscores the significance of PAR structure in protein binding, enzymatic degradation, and phase separation processes, with implications for downstream signaling pathways and biomolecular condensate formation. Despite advances, the functional nuances of PAR chain diversity and its interplay with other cellular mechanisms remain areas of active investigation, promising novel therapeutic insights through targeted modulation of ADP-ribosyltransferase (ART) activities.
Classifications and functions of ARTs
PARylation is carried out by a large family of ADP-ribosyltransferases (ARTs), also known as poly-ADP-ribose polymerases (PARPs).2–4 ARTs are present across all domains of life and carry crucial functions in many cellular processes including DNA repair, cellular transport, transcription regulation, and cell death.5,6 In humans alone, 17 ARTs have been identified to date. 4
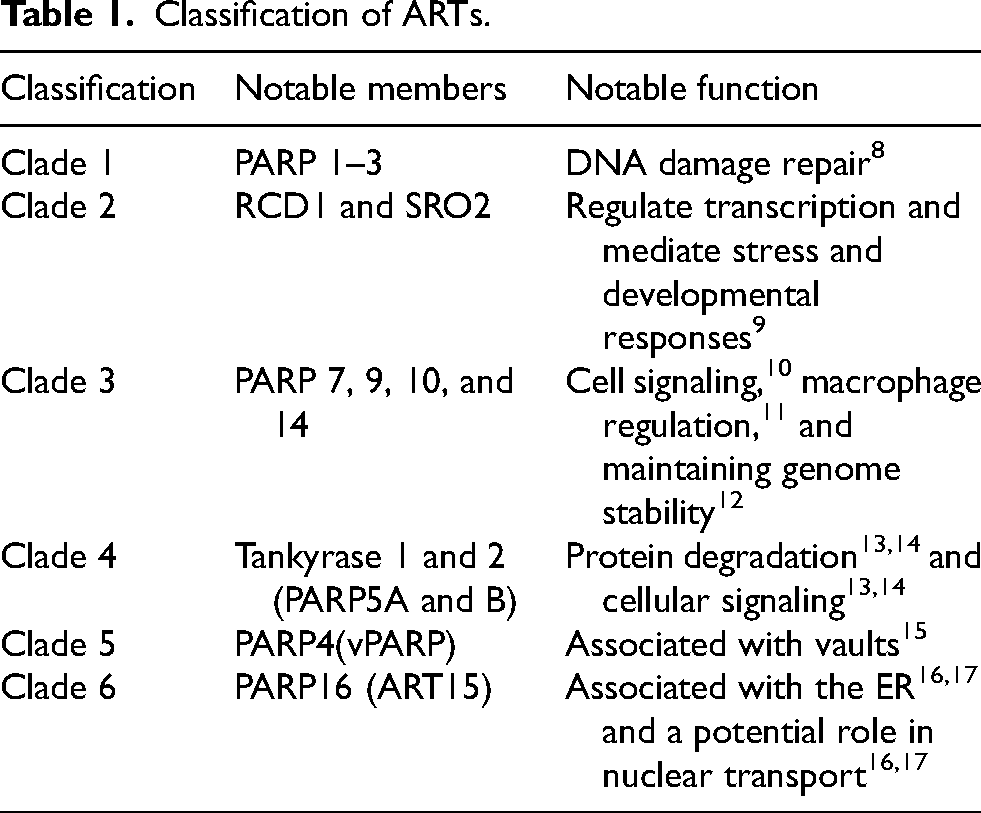
Based on evolution, eukaryotic ARTs can be classified into six clades (Table 1). 7 Clade 1 consists of many PARylating proteins involved in DNA damage repair, and some also play a role in chromatin and transcription regulation. 5 Notable examples of clade 1 ARTs include human PARPs 1–3, which possess DNA-binding domains and are activated by DNA damage. 8 Clade 2 consists of a distinct class of ARTs only present in plants. Examples include RCD1 and SRO2, which regulate transcription and mediate stress and developmental responses. 9 Clade 3 consists of a heterogeneous group of ARTs that varies greatly in structure and function. 7 Human PARPs 7(TIPARP), 9, 10, and 14 are members of the clade with diverse functions including cell signaling, macrophage regulation, and maintaining genome stability.7,10–12 Clade 4 ARTs have a distinctive domain structure consisting of multiple ankyrin repeats followed by a sterile alpha motif. 7 Human PARPs in the clade include tankyrase 1 and 2 (PARP5A and B) and play a role in protein degradation and cellular signaling.13,14 Clade 5 is characterized by the position of the catalytic domain in the middle of the protein. In humans, PARP4 (vPARP) has been associated with vaults, the largest ribonucleoprotein particles with unknown function.15,18 Clade 6 is characterized by its catalytic domain and most of its members have not been characterized functionally with no known functional domain at the N terminal region. 7 Human PARP16 (ART15) however is one of the exceptions, and studies have shown its involvement with the endoplasmic reticulum and a potential role in nuclear transport.16,17 Given the emerging roles of defective DNA damage repair (DDR) in neurodegenerative diseases, we will focus on ARTs involved in DDR.
Classification of ARTs.
PARP1 in DNA damage repair
Several ARTs are involved in DNA damage repair, and PARPs 1–3 in particular are DNA-dependent. 8 PARP1 is the largest and most extensively studied ART of the three due to its abundance in cells and its involvement in a wide range of DNA damage repair processes. 8 PARP1 is thought to carry out most of the PARylation in the cell and plays an important role in DNA damage repair. 19 It rapidly recognizes single and double-stranded DNA damage breaks through its DNA binding domain and PARylates itself and other DNA damage repair proteins to recruit and activate them. 8 PARP1 knockout mice are highly susceptible to DNA damaging agents, and PARP1 and PARP2 knockout results in embryonic lethality.20,21
A well-characterized involvement of ARTs in DNA damage repair is their role in single-stranded break (SSB) repair.22,23 PARP1 and 2 rapidly detect the presence of single-strand breaks and initiate the recruitment of XRCC1, DNA ligase III, and other proteins crucial to the repair process.24,25 PARP1 also recognizes apurinic/apyrimidinic sites but its exact involvement in base excision repair has been an area of debate.23,26
Double-stranded breaks (DSB) are repaired through two main repair pathways, homologous recombination (HR) and non-homologous end-joining (NHEJ), which further divides into alternative NHEJ and classic NHEJ.27,28 PARP1 plays an important role in DSB repair through the recognition of DNA damage, recruitment of the repair complex, and determination of the repair pathways. 23 PARP1 and Ku compete for binding to exposed DNA ends at the site of damage to determine which DSB repair pathway will take place.29–31 During the repair process, PARP1 also recruits the MRN complex and activates ATM, an important signaling protein for DSB repair.32,33
An essential event for DNA damage repair that occurs concurrently with the repair process is the relaxation of chromatin to allow the repair complex to access DNA. 34 The process is regulated by many post-translational modifications including PARylation, and PARP1 has been shown to PARylate all histone subunits and the linker histone H1.35,36 PARP1 also recruits other chromatin remodeling proteins such as histone PARylation factor 1, which further promotes histone PARylation. 37 PARP1 also indirectly assists in DNA damage repair through replication fork stalling. 38 During DNA replication, the replication fork may encounter damaged DNA. Stalling the replication process and efficient repair of the damaged DNA is crucial to prevent errors in DNA replication and maintain genome integrity. PARP1 is activated by the stalled replication forks, promotes HR at the sites of damage, and restarts DNA replication after the repair. 38
PARP1 is cleaved by caspases, which does not affect PARP1 activity, but does implicate PARP1 in disease mechanisms.39,40
PAR dysregulation in neurodegenerative disease
Neurodegenerative diseases are a broad spectrum of disorders with varying clinical presentations and underlying causes of disease. 41 They are characterized by progressive loss of neurons and impairment of cognitive and motor functions. 41 Although the precise disease mechanisms remain unknown, evidence suggests they are affected by factors including DNA damage, oxidative stress, mitochondrial impairment, and neuroinflammation.42,43
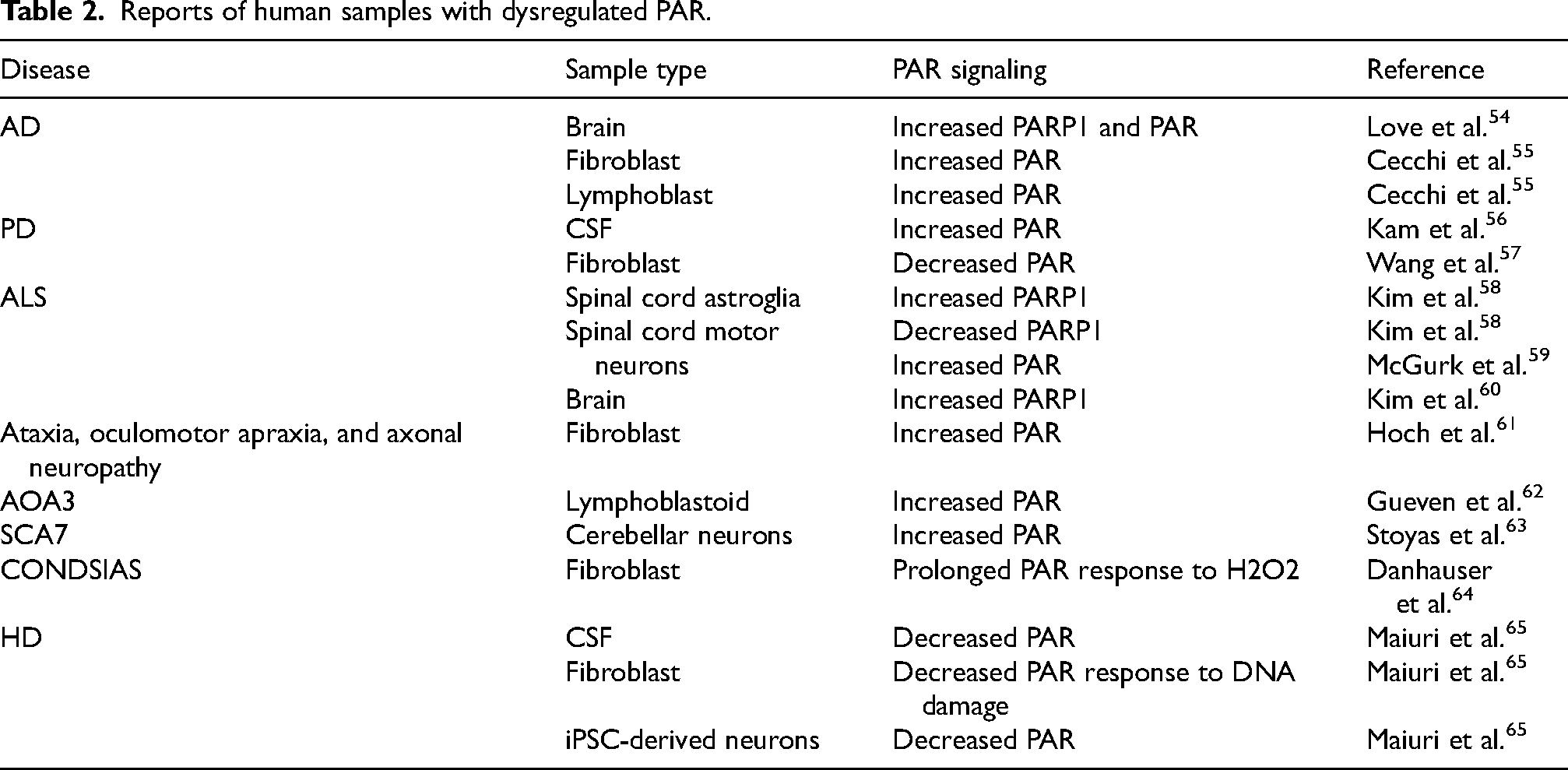
PARylation has been implicated in many of these mechanisms. As previously mentioned, PARylation plays an important role in DNA damage signaling, recruitment of repair proteins, and acts as a scaffold in the repair process. 44 PARP1 promotes DNA damage repair caused by oxidative stress, and is involved in the removal of oxidized proteins, serving a protective function against oxidative stress.45,46 Alternatively, PARylation is an energy demanding process due to the consumption of NAD+, and its overactivation can result in severe NAD+ depletion, mitochondrial dysfunction, and eventual cell death.47–50 Aberrant PAR signaling has also been observed in multiple neurodegenerative diseases, including Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, cerebellar ataxias, and Huntington disease,42,51–53 which we will summarize in turn, with emphasis placed on evidence from human samples (Table 2).
Reports of human samples with dysregulated PAR.
Alzheimer disease (AD)
Aberrant PAR signaling has been observed in AD for over two decades, and subsequent studies have since provided further support for this link.54,55,66,67 Elevated PARP1 and PAR levels were observed in the AD brain compared to control, 54 and higher levels of PARP1 PARylation were observed in AD fibroblasts and lymphoblasts, without changes in overall PARP1 levels. 55 Genetic analyses have also linked the PARP1 gene to AD susceptibility,67,68 further highlighting the potential role for PAR in AD pathogenesis. However, increased PAR levels are only correlative with disease, which is a concern in many neurodegenerative diseases. Given that most of these diseases are late age onset with increased reactive oxygen species (ROS) loads, it is likely elevated PAR levels are due to elevated DNA damage. Indeed, in animal models, both olfactory and hearing loss in AD models are correlated with high PAR levels and low NAD+ levels.69,70
Parkinson disease (PD)
The presynaptic neuronal protein, α-synuclein, has long been associated with PD. 71 More recently, the activation of PARP1 was shown to exacerbate α-synuclein toxicity. 56 Further, preformed fibrils of recombinant α-synuclein induced PARP1 activation, resulting in α-synuclein aggregates and cell death via parthanatos, a non-apoptotic mechanism involving PAR and apoptosis-inducing factor (AIF). 72 The use of PARP inhibitors was able to rescue toxicity induced by the α-synuclein preformed fibrils.
In the context of 6-OHDA and MPTP mouse models, which use neurotoxins to induce PD-like symptoms, PARP1 knockout was protective against neuronal death.73,74 In the MPTP mouse model, PARP1 overactivation through oxidative stress also resulted in energy depletion, mitochondrial depolarization and AIF translocation. 75 While the vast majority of evidence for a link between PAR and PD comes from mouse and in vitro studies, elevated PAR levels were found in the CSF of PD patients, 56 and interactions between PAR and phosphorylated α-synuclein were detected in postmortem PD patient samples. 76 As discussed below, dysregulated PAR signaling was also seen in PD patient fibroblasts. 57 As with AD, this may just be indicative of more DNA damage but in these diseases, a threshold may be crossed that nets neuronal loss by parthanatos.
Amyotrophic lateral sclerosis (ALS)
ALS affects motor neurons in the brain and spinal cord, which normally control voluntary movement and breathing. In one study investigating spinal cord tissue from ALS patients, Kim et al. found PARP1 immunoreactivity to be increased in astroglia, but reduced in spinal motor neurons. 58 The same authors later investigated ALS brain tissue and found increased PARP1 expression in the neurons, subcortical glia, and macrophages of the motor cortex, as well as the parietal cortex and cerebellum, regions that are not typically affected in ALS. 60 In contrast to the findings of Kim et al., McGurk et al., found that the motor neurons in ALS spinal cord exhibited elevated PAR. 59 This could be explained by higher PARP1 activity despite lower levels, similar to what was seen in AD patient fibroblasts and lymphoblasts. 55 The mechanism by which elevated PAR signaling is connected to ALS remains unclear, but could be mediated through ALS-associated proteins TDP-43, FUS, and C9orf72, all of which can be regulated by PARP1 and PAR.77–80 Of the neurodegenerative diseases discussed here, ALS has the clearest connection to DNA damage as the result of a higher ROS load, with several known mutations in superoxide dismutase (SOD1). 81
Cerebellar ataxia
Cerebellar ataxia is a collection of movement disorders characterized by dysfunctions related to the cerebellum, resulting in the impaired control of limb and ocular movement, balance, and gait. 82 Defective DNA damage repair and PARP dysregulation have been implicated in several cerebellar ataxias. 83 In one case, a patient carrying mutations in the DNA repair gene XRCC1 was diagnosed with symptoms of cerebellar ataxia, and the disease pathology was associated with hyperactivation of PARP1. 61 Fibroblasts derived from the patient had increased levels of PAR, and loss of cerebellar neurons in a Xrcc1-defective mouse model was rescued by deletion of Parp1. 61 Elevated PAR levels were also observed in lymphoblastoid cells from a patient with ataxia oculomotor apraxia type 3(AOA3), a rare inherited disorder characterized by hypersensitivity to oxidative stress and apoptosis resistance. 62
Spinocerebellar ataxia type 7 (SCA7) is an autosomal dominant disorder characterized by progressive cerebellar degeneration and motor incoordination, and elevated levels of PAR were observed in cerebellar neurons of SCA7 patients. 63 In SCA7 mouse models, NAD+ depletion was responsible for dysregulation of Sirt1, a NAD+ -dependent deacetylase. The authors therefore propose that excessive PARP1 activity may be responsible for the NAD+ depletion and neuronal demise. 63 This mechanism could be relevant to other diseases with elevated PAR levels, as PARP1 activity can metabolically drain neurons of NAD+ leading to a severe stress of energy crisis. 84 In a highly metabolically active cell population, this may in part explain the brain pathology predominant in these diseases.
Elevated PAR levels may arise not only by excessive polymerization, but also by deficient hydrolysis. Indeed, pathogenic variants of ADPRHL2, encoding the mono(ADP-ribosyl) hydrolase ARH3, have been found in individuals exhibiting neurodegenerative symptoms including cerebellar atrophy and progressive ataxia, a disorder now termed childhood-onset neurodegeneration, stress-induced, with variable ataxia and seizures (CONDSIAS) (see Bannister et al. 85 and references therein). One of the earliest reports of ataxia-associated ARH3 deficiency showed a prolonged PAR response to H2O2 in patient fibroblasts. 64
It is worth noting that some ataxias have been associated with normal PARP1 activity. 86 This could be because the ataxia is caused by another mechanism, or, in the context of elevated DNA damage and oxidative stress, 86 this could be an indication of deficient PAR signaling. As discussed below, insufficient PAR signaling may also result in neuronal death, providing an alternate pathogenic mechanism in neurodegenerative disease.
Thus, most of the accumulating evidence linking PAR dysregulation with neurodegenerative disease points to increased PARP1 levels or activity, leading to increased PAR levels (Table 2). As neurons are highly energy dependent, excessive production of PAR could be detrimental by depleting NAD+ stores 47 or by blocking glycolysis via hexokinase inhibition. 87 It could also lead to the translocation of AIF from the mitochondria to the nucleus, resulting in cell death by parthanatos. 52 However, we propose that a decrease in PAR signaling could also lead to neuronal death, based on our findings in samples from people with Huntington disease.
PAR dysregulation in Huntington disease
Although minimal, previous evidence supported a role for increased PAR signaling in Huntington disease, like other neurodegenerative diseases. A classic study has often been cited as an example of increased PARP1 in HD brain, however they measured the 85-kDa caspase-cleaved fragment (p85) of human PARP as a marker for apoptosis, 88 which differs from active PAR-generating PARP1. 89 Two more studies described the beneficial effects of PARP1 inhibition in the R6/2 mouse model of HD.90,91 In contrast, we recently found decreased PAR levels in the CSF of HD patients, both at pre-manifest and manifest stages. 65 We further showed that the PAR response to DNA damage was impaired in HD patient-derived fibroblasts and neurons differentiated from iPSCs. At the molecular level, we found that both wild type and mutant huntingtin protein could bind PAR directly, but only the wild type protein was capable of stimulating PARP1 activity in vitro. This biochemical data was reinforced by direct molecular imaging using atomic force microscopy, where it was seen that pure, recombinant huntingtin/HAP40 dimer could be seen binding to -ve ends of growing PAR chains at the single molecule level. This provides a compelling potential mechanism in which the normal huntingtin protein functions to promote PARP1 activity, but this function is impaired in HD.
While these results are at odds with the previous reports in mice,90,91 it is possible that the R6/2 mouse model, which expresses only the first 81 amino acids of the 3144-amino acid huntingtin protein, may not reflect the normal function of full-length huntingtin in PAR biology, and therefore exhibit different phenotypes than patient-derived materials. Indeed, the defined PAR-binding motif in huntingtin is between residues 1790–1798, which demonstrates that this interaction cannot occur in a small amino-terminal sub fragment of huntingtin. PAR levels in those studies were not measured, so we do not know whether PAR levels were elevated in HD mice, nor whether they were decreased after treatment with PARP inhibitor. It is also possible that PARP1 inhibition was beneficial in mice (and would in fact be beneficial in HD patients), if the “treadmilling” activity of PARP1, 92 by which PARP1 hydrolyzes NAD+ to generate free ADP-ribose, is responsible for the reduced PAR levels that we observed. 65 Alternatively, the results could be explained by yet another mechanism, as is the case in a mouse model of spinal and bulbar muscular atrophy (SBMA), which benefited from olaparib treatment, despite having lower levels of PAR in quadricep tissue, via restoration of hexokinase activity and glycolysis. 93
Huntingtin is not the only neurodegenerative disease-associated protein that has been reported to promote PARP1 activity: DJ-1 is a causative gene of familial Parkinson disease (PD) 94 that regulates DNA repair through the PARP1 pathway. 57 Like huntingtin, the wild-type DJ-1 protein can stimulate PARP1 activity in vitro, and similar to our findings in HD fibroblasts, cells from PD patients with the DJ-1 mutation also have defective PARP1 activity and impaired repair of DSBs. 57 This is in contrast to the increased PAR observed in PD CSF samples, 56 cases which may have arisen sporadically, or by mutations in other genes. It can therefore be hypothesized that the net result of dysregulated PAR, whether increased or decreased, is a similar outcome of neuronal death (Figure 1).
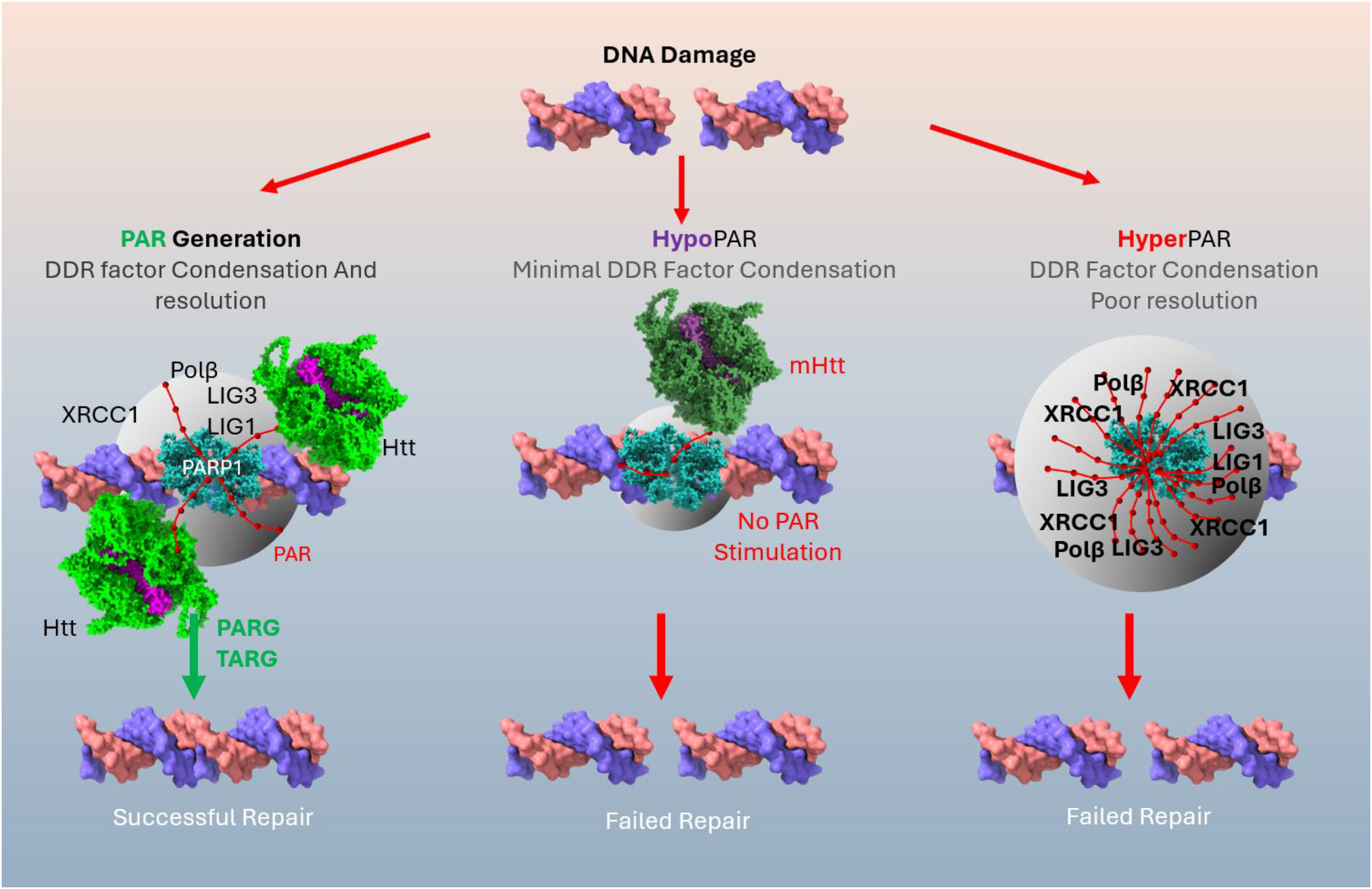
Balanced PAR signaling is required for neuronal health, while tipping the equilibrium in either direction can lead to neuronal death. PAR generation, stimulated by huntingtin, recruits DNA repair factors to DNA breaks and triggers condensation of repair factors, regulated by PAR and resolved by glycosylases PARG and TARG. HypoPARylation by failure of mutant huntingtin to stimulate PARP1 results in poor repair due to lack of proper protein recruitment. HyperPAR complexes get trapped on DNA, sequestering cell NAD+ levels lowering energy and resulting in failed repair.
Deficient PAR signaling may lead to the elevated DNA damage seen in HD models and patient samples, including the zQ175 mouse striatum95,96 and mouse embryo fibroblasts from the BACHD mouse, 97 HD patient-derived fibroblasts65,98–100 and lymphocyte cell lines, 101 human embryonic stem cell-derived CNS and peripheral cells 102 and induced pluripotent cell-derived striatal neurons95,103 and astrocytes, 104 peripheral blood mononuclear cells,105,106 and post-mortem human brain. 97 In turn, excess DNA damage could explain mitochondrial dysfunction,107,108 energy depletion,109,110 neuroinflammation,111,112 dysregulated autophagy,113,114 and protein aggregation,115,116 all of which are classical observations in the field.
Recent biophysical studies on the role of PARylation and DNA repair identify a mechanism of PAR chains inducing and regulating the condensation of proteins into a liquid-liquid phase separated state. 117 This phase separation is regulated by PAR. 118 Biochemical and cell biology studies have identified huntingtin and polyglutamine containing huntingtin fragments as able to undergo liquid-liquid phase separation119,120 (Figure 1).
Given the pleiotropic role PARPs in with least 800–1000 human proteins known to be modified by PARP-mediated ADP-ribosylation. This landscape could be changed in HD and defined using quantitative mass spectrometry in cellular and in vivo models of HD, or from human HD brains.
PARylation and HD GWAS
Recent genome-wide association studies (GWAS) in HD have identified DNA ligase I (LIG1) as a modifier of disease onset. 121 Multiple proteins involved in the mismatch repair (MMR) pathway, including MSH3, FAN1, and MLH1, have also been implicated. LIG1 plays an important role in MMR through the ligation of nicked DNA caused by the repair process. LIG1 and PARP1 have complementary functions in several DNA repair pathways, which implicates LIG1 as a sensitizer of PARP1 inhibitors. 122 Similarly, the combined dysregulation of PAR signaling and LIG1 variants in HD could further contribute to HD pathology.122,123
Within HD brains, LIG1 protein was seen in neurofibrillary tangles along with RRM2B or P53R2, another HD GWAS modifier, 124 suggesting a possible mechanistic connection. While misnamed Ribonucleotide Reductase M2B, RRM2B protein is not an enzyme, it is the regulatory subunit of the ribonucleotide reductase (RNR) complex. In human disease, RRM2B mutants lead to a rare syndrome of mitochondrial DNA depletion,125,126 as well as a variety of rare disorders. 127 The RNR complex can salvage ribonucleotides, such as poly-ADP ribose from transient PAR chains in conjunction with thioredoxin scaffolded on RRM2B. 128 The thioredoxin pathway is a fundamental cellular system responsible for maintaining redox homeostasis by facilitating the reduction of disulfide bonds in proteins. As with PARylation, thioredoxin activity is fuelled by NAD+ . Thus with LIG1 and RRM2B, there is a crossover of regulation between cell metabolism, NAD+ levels and PARylation. As a synthetic lethal in BRCA1 breast cancer tumors, LIG1 inactivation results in increased PARylation. 129
Like huntingtin, RRM2B is regulated by TP53. 130 Ergo, any cell based studies of RRM2B need to avoid transformed cell lines in which TP53 pathways are typically inhibited. 131 Deficiency in TP53/RRM2B results in the activation of an NRF2 antioxidant transcriptional program, with a concomitant elevation in basal PARylation in cells. 131 As a corollary, RRM2B suppresses activation of the oxidative stress pathway and is up-regulated by TP53 during senescence. 132 The effect of NRF2 on elevating the PAR response may have therapeutic implications in Huntington disease with modern NRF2 stabilizing drugs like omaveloxolone (Skyclarys), recently approved for Friedreich's ataxia. 133
Outlook
Here, we have summarized the reports of dysregulated PAR signaling in human neurodegenerative disease samples. While many conditions, such as Alzheimer, Parkinson, ALS, and ataxia, are predominantly characterized by elevated PAR levels and PARP1 overactivation, Huntington disease presents a striking exception, with reduced PAR levels and impaired PARP1 activity. In the human biology context, Huntington disease gene carriers have a significantly reduced incidence of some cancer types, consistent with the success of PARP1 inhibitor drugs in oncology applications. It could be important to know outcomes of any HD patients treated at any time in their lives with PARP1 inhibitors and how this affected cancer treatment or had any effect on HD symptoms. The widespread ethnic incidence of HD across the world 134 suggests that a mutant huntingtin gene may have conferred a genetic advantage in evolution to avoid early life cancers and thus a better chance of procreation.
We present a model in which balanced PAR signaling is required for neuronal health, while tipping the equilibrium in either direction can lead to neuronal death. This diversity in PAR dysregulation highlights the need to consider disease-specific profiles of PAR signaling when developing therapeutic interventions. Moving forward, it will be crucial to explore both PARP inhibitors and PARG inhibitors as potential treatments, tailoring their use to the unique signaling patterns observed in each disorder. Broader efforts to identify cases of reduced PAR signaling and elucidate their underlying mechanisms will provide valuable insights, paving the way for novel therapeutic strategies and a more nuanced understanding of PAR biology in neurodegeneration.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by operating grants to R.T. from the Krembil Foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.