Parkinson's disease (PD) is characterized by progressive motor and non-motor dysfunction arising from synaptic pathology that precedes neuronal loss. Synaptic dysfunction represents a central pathological mechanism in both familial and sporadic PD, with multiple PD-linked genes directly regulating synaptic vesicle (SV) trafficking. Here, we review recent studies linking SV dysfunction to PD pathology by examining how disease-linked proteins dysregulate distinct steps in the SV cycle and by exploring their cascading effects on synaptic physiology and circuit function. We highlight three critical pathogenic mechanisms: (1) impaired neurotransmitter import and storage, (2) disrupted SV pool organization, and (3) altered SV exocytosis and endocytosis. We also review recent studies investigating how presynaptic pathology triggers impairments in postsynaptic plasticity and circuit-level reorganization across brain regions. Together, these studies highlight that presynaptic SV dysfunction is a central mechanism in PD pathogenesis and therefore a promising target for developing therapeutic strategies aimed at reverting these early disease-driving synaptic changes.
Plain language title
New insights into synaptic vesicle dysfunction in Parkinson's disease
Plain language summary
The earliest sign of both inherited and sporadic forms of Parkinson's disease (PD) is damage to nerve cell endings called synapses. Synapses are connections between nerve cells that mediate communication between these cells. Synaptic vesicles are tiny packages inside synapses that store and release chemical messengers called neurotransmitters. Neurotransmitter release activates neighboring nerve cells to propagate the signal. Storage, release and recycling of synaptic vesicles is a precisely regulated process and involves many proteins and lipids. When this process malfunctions, synapses fail, triggering a pathological cascade throughout the brain. Here, we review recent findings in the field that demonstrate how dysfunctional proteins and lipids disrupt normal synaptic vesicle function and thereby damage synapses and brain networks. We focus on three major mechanisms driving PD: (1) dysfunctional import and storage of neurotransmitters in synaptic vesicles; (2) defective transport of synaptic vesicles to synapses; and (3) breakdown of the recycling system that releases and retrieves synaptic vesicles. Our review highlights that many key proteins involved in these processes become dysregulated in PD, some of which are already genetically linked to PD. Synaptic vesicle dysfunction then triggers downstream problems, causing receptors on the signal-receiving neighboring nerve cells to lose their sensitivity and triggering abnormal reorganization of brain networks. These changes spread across interconnected brain regions, producing the diverse symptoms in PD such as changes in movement, mood, and cognition. Together, these studies highlight that synaptic damage is a central mechanism in PD and therefore a promising target for developing therapeutic strategies aimed at early stages in the disease process.
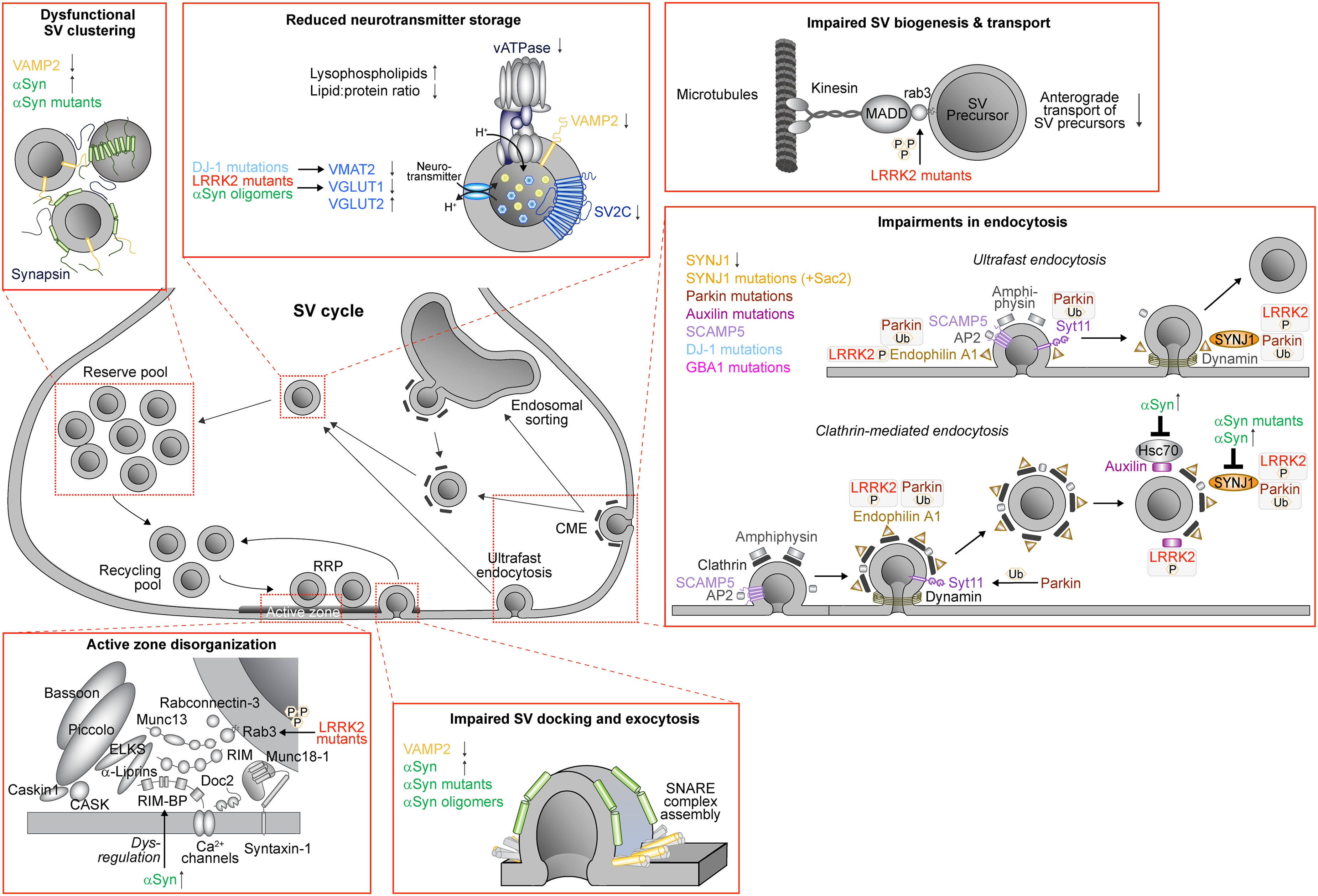
Parkinson's disease (PD) is the second most common neurodegenerative disorder and characterized by motor dysfunction.1 Frequently, non-motor symptoms such as REM sleep behavior disorder (RBD), depression, anxiety, anosmia, constipation, and cognitive decline are also observed.1 Mitochondrial and lysosomal dysfunction have long been implicated in PD pathology. Alterations in PARK2/PRKN (parkin), PARK6 (PINK1), PARK7 (DJ-1), PARK15 (FBX07), PARK17 (VPS35), and PARK23 (VPS13C) lead to impairments in complex I function, mitophagy, mitochondrial fission and fusion, and mitochondrial biogenesis.2 Autophagy-lysosomal impairments are genetically linked to PARK2/PRKN (parkin), PARK6 (PINK1), PARK8 (LRRK2), PARK9 (ATP13A2), PARK17 (VPS35), GBA1 (glucocerebrosidase 1), and TMEM175 (transmembrane protein 175).3 More recently, cumulative evidence points to synaptic dysfunction as a central pathological mechanism in both familial and sporadic PD which emerges months to years before motor symptom onset and neuronal loss.4 Central to this process is the synaptic vesicle (SV) cycle. SVs are small presynaptic organelles that store and release neurotransmitters in a series of well-regulated steps.5 They are composed of a lipid bilayer and are densely packed with proteins that regulate SV transport, SV pools, neurotransmitter import, SV exocytosis and endocytosis.6–8 Several of these processes are dysregulated in PD, including SV biogenesis, neurotransmitter storage and SV clustering,5,8,9 which triggers postsynaptic and circuit-level impairments.10 Noteworthy, many of the genes linked to familial PD are directly or indirectly involved in the SV cycle, including PARK1/PARK4/SNCA (α-synuclein), PARK2/PRKN (parkin), PARK6 (PINK1), PARK7 (DJ-1), PARK8 (LRRK2), and GBA1 (GBA), pointing to SV dysfunction as a central pathological mechanism in PD. Here, we focus on recent advances that link SV dysfunction to PD pathology by reviewing the impact of PD-linked SV proteins on distinct steps in the SV cycle and associated outcomes on synapse and circuit functionality (Figure 1 and Table 1).
Effects of PD-linked synaptic vesicle proteins and lipids on the SV cycle. Overview of the SV cycle in presynaptic nerve terminals that highlights SV pools, SV exocytosis at the active zone and different routes of endocytosis. In PD, every step of the SV cycle is affected. Insets highlight these steps and include effects on neurotransmitter import and storage in SVs, clustering of the reserve pool of SVs, organization of the active zone, docking and exocytosis of SVs, and endocytosis of SVs (RRP, readily-releasable pool; CME, clathrin-mediated endocytosis). Highlighted are proteins discussed in this review.
Summary of PD-associated presynaptic proteins and lipids that are altered in PD patient tissue and/or in PD animal models. References are provided in the main text.
Protein (PARK gene)
PD-associated changes
α-Synuclein (PARK1/PARK4)
PD-linked mutations and overexpression shift αSyn towards toxic oligomerization and aggregation, impairing SV trafficking, clathrin-mediated endocytosis, and SNARE-complex assembly, which leads to synaptic dysfunction, dopamine neuron degeneration, and progressive circuit-level dopamine/GABA imbalance that drives motor and non-motor symptoms of PD.
Auxilin (PARK19)
Auxilin variants lead to increased levels of GAK and reduced levels of tyrosine hydroxylase, DAT and VMAT in human CSF. Auxilin deficiency impairs uncoating of clathrin-coated vesicles and thereby leads to their accumulation in synaptic terminals, and causes SV trafficking defects, reduced SV numbers, cytoplasmic dopamine accumulation and oxidation, leading to PD-like motor deficits.
DJ-1 (PARK7)
DJ-1 deficiency impairs SV endocytosis and VMAT2-mediated dopamine storage in SVs, and causes prolonged exocytosis, cytoplasmic dopamine oxidation, aberrant SV morphology, and motor deficits with dopaminergic neurodegeneration.
GBA
GBA variants impair SV trafficking and synapse assembly, reducing SV and clathrin-coated vesicle numbers. GBA dysfunction produces variant-specific alterations in presynaptic function in human-derived dopaminergic neurons and, when combined with αSyn dysfunction, causes cognitive and motor decline.
LRRK2 (PARK8)
LRRK2 mutations lead to aberrant protein phosphorylation and impair trafficking of SVs and their precursors, leading to presynaptic dysfunction at multiple levels.
Parkin (PARK2)
PD-linked mutations cause neuron-type and circuit-specific presynaptic deficits via impaired protein ubiquitination which leads to dysregulated endocytosis.
SCAMP5
Reduction of SCAMP5 levels leads to elevated αSyn levels and αSyn oligomerization, SV hyperacidification, deficient dopamine secretion, dopamine dyshomeostasis, degeneration of dopaminergic neurons, and PD-like motor phenotypes.
SV2C
SV2C contributes to dopamine homeostasis by assisting in retaining neurotransmitters within SVs, and contributes to MPTP resistance, suggesting that reduced SV2C levels in PD patients may impact specifically dopaminergic neuron function.
Synaptojanin-1 (PARK20)
Reduced levels and variants impair endocytosis via dysfunctional PIP dephosphorylation and promote αSyn aggregation, while its cleavage causes dopaminergic neurodegeneration and PD pathology.
Synaptotagmin-11
Synaptotagmin-11 knockout enhances SV endocytosis, whereas its overexpression impairs evoked dopamine release and induces motor and depressive-like behaviors typical for PD.
VAMP2/synaptobrevin-2
VAMP2 is essential for neurotransmitter release and shows PD stage-dependent changes. Reduced levels in patient extracellular vesicles and late-stage PD brain tissue likely reflect synaptic failure.
vATPase
Dysregulation of the vATPase is expected to result in deficits in neurotransmitter storage in SVs due to generating an insufficient electrochemical gradient to drive neurotransmitter import into SVs.
VGLUT2
VGLUT2 supports resilience of dopaminergic neurons, and higher levels in females may explain lower PD prevalence in women.
VMAT2
Reduced levels lead to a reduction in SV dopamine content and accumulation of cytosolic dopamine, resulting in accumulation of dopamine-derived oxidative species which causes impairments in dopaminergic neurotransmission and PD symptomology.
Lipids
PD-associated changes
Glycerolipids
PD patients carrying G2019S mutant LRRK2 display altered glycerolipid metabolism, which may contribute to dysregulation of SV trafficking.
Lysophospholipids
Elevated levels of lysophospholipids observed in a murine PD model may lead to altered SV curvature, packing defects, and clustering, along with impaired SV fusion and fission kinetics.
Phospholipids
Phospholipids serve as structural components of membranes and determine their biophysical properties. They are also binding hubs for proteins and involved in signal transduction. Changes in phospholipid levels may alter fluidity, shape and integrity of SV membranes and affect interactions with peripherally associated SV proteins.
Phosphatidylinositol
PI(3,4,5)P3 accumulates in human PD brain and colocalizes with phosphorylated αSyn. Synaptojanin-1 dysfunction elevates PI(3,4,5)P3 and PI(4,5)P2 levels. GBA variants lead to reduced PI(4,5)P2 levels. Auxilin variants result in reduced levels of PI and lipids containing polyunsaturated fatty acids (PUFAs). Changes in PIP metabolism result in impaired cell signaling, SV trafficking, dysfunctional membrane organization, and αSyn aggregation.
Sphingolipids
PD patients carrying G2019S mutant LRRK2 or juvenile parkinsonism patients carrying K295R mutant SYNJ1 present with altered sphingolipid metabolism. Imbalances in sphingolipids lead to disrupted SV trafficking, reduced neuroprotection, and impaired autophagy.
Lipid:protein ratio
Decreased lipid:protein ratio on SVs in a murine model of PD may lead to altered SV curvature and excessive molecular crowding of proteins on the SV surface. These changes are expected to affect biophysical membrane properties and protein-membrane interactions leading to impaired SV trafficking.
Synaptic vesicle composition and biogenesis
SV proteins are synthesized in the soma and transported as immature precursor vesicles. The mechanisms that generate SVs from precursor vesicles remain unclear and include maturation along their transport route and/or upon arrival at presynaptic terminals.9 An important morphological feature of SVs is their uniform size, which ensures that well-defined amounts of neurotransmitters are stored in each SV. Quality control mechanisms exist to ensure the formation of functional SVs with specific lipids and proteins. Defects in SV biogenesis and recycling are linked to PD and involve SV proteins and lipids.11–13 A recent study in mice identified proteomic and lipidomic alterations that are expected to affect SV pools, exocytosis and endocytosis, which include elevated lysophospholipid levels, reduced lipid:protein ratio, elevated levels of the endocytic proteins AP1B1, Bin1, PICALM and synaptojanin-1, and of exocytosis- and SV pool-regulating proteins,8 some of which are genetically linked to PD.
Neurotransmitter import and storage
Accurate neurotransmitter import and storage are critical because they maintain adequate SV filling to sustain synaptic transmission, prevent oxidative damage from cytoplasmic neurotransmitter accumulation, and enable synaptic plasticity.14 Neurotransmitter import and storage dysfunction is observed in both PD patient tissue and preclinical models. Although PD is frequently referred to as a dopamine storage disorder, other neurotransmitters contribute to PD pathology as well but are understudied and many times overlooked. Recent studies highlight the involvement of dysfunctional glutamatergic signaling and general neurotransmitter storage deficits, as discussed in detail below.
vATPase: Vacuolar-type adenosine triphosphatase (vATPase) is crucial for neurotransmission.15 The 16 subunit16 proton pump acidifies SVs to ∼pH 5.7,17 generating an electrochemical gradient that is utilized by SV neurotransmitter transporters to import neurotransmitters. In a PD mouse model, proton-pumping V0 subunits (a1 and d1), and the ATP-hydrolyzing V1 subunit (F1) are dysregulated.8 Intriguingly, the V0 domain forms a complex with the SNARE protein VAMP2 and synaptophysin, which may regulate vATPase copy number on SVs.18,19 Luminal SV phospholipids (PC, PE, PS) are involved in stabilizing the core of the V0 domain and perhaps its assembly,20 and are dysregulated with age and as a function of α-synuclein (αSyn).8
VMAT2 (SLC18A2): Vesicular monoamine transporter VMAT2 loads SVs with dopamine, serotonin, histamine, adrenaline, and noradrenaline. VMAT2 expression levels are reduced in PD, dementia with Lewy bodies (DLB), and RBD patients.21–24 Loss of function variants cause infantile parkinsonism-dystonia 2,25 while gain-of-function haplotypes are PD-protective, specifically for females.26 Reduced VMAT2 levels drive dopamine storage dysfunction and PD. VMAT2 knockout zebrafish develop PD-like motor, anxiety and sleep disorder phenotypes with reduced number of tyrosine hydroxylase-positive neurons, which is rescued with dopamine agonists and precursors.27 VMAT2 knockdown in rats causes cytoplasmic dopamine accumulation, followed by degeneration of dopaminergic neurons and a PD-like motor phenotype, and accompanied by LRRK2 activation, rab10 phosphorylation and αSyn pathology.28 VMAT2 overexpression in humanized rats reduces dopamine oxidation and attenuates PD-like pathology.29 In MPTP-treated mice, HDAC inhibitors increase VMAT2 levels, reduce dopamine-induced oxidative stress and improve motor behavior, which is further enhanced by L-DOPA co-administration.30 Inhibitors also result in increased dopamine levels in synaptosomes isolated from SH-SY5Y cells.30 6-OHDA-treated SH-SY5Y cells develop αSyn accumulation with decreased levels of dopamine and tyrosine hydroxylase.31 In MPTP-treated mice, the glucoside gastrodin reverses motor symptoms, increases tyrosine hydroxylase and VMAT2 levels, restores SV size and numbers, and rescues dopamine levels. Gastrodin also restores dopamine levels and decreases ROS levels in MPTP-treated PC12 cells.32
VGLUT: Vesicular glutamate transporters (VGLUT) load SVs with glutamate, which is co-released with dopamine in the substantia nigra pars compacta,33 although not necessarily from the same vesicle.34 VGLUT1 is primarily expressed in the cerebral cortex, hippocampus, and cerebellum. VGLUT2 is present in subcortical structures, such as the thalamus, amygdala, hypothalamus, and brainstem.35–37 Analysis of postmortem cortical PD brain tissue revealed hypomethylation in VGLUT2 gene in females.38 Surviving dopaminergic neurons in PD patients have higher VGLUT2 mRNA levels,39 suggesting that VGLUT2 provides resilience, supported by studies in rodents40,41 and iPSC-derived human cortical glutamatergic neurons.42 VGLUT1 shows opposite effects, where injection of αSyn preformed fibrils (PFF) into the striatum triggers loss of intracortical VGLUT1-positive excitatory synapses.43 VGLUT may be a driver for higher PD prevalence in males.44 Female flies, rodents and humans have higher VGLUT2 levels. Aging male flies show stronger motor dysfunction than females, where age-dependent motor dysfunction could be triggered with VGLUT knockdown.45
Synaptic vesicle pools and exocytosis
Proper coordination of SV pools and exocytosis is essential because it sustains neurotransmitter release during activity, gates short-term plasticity, and maintains the fidelity of synaptic signaling. Impaired SV pool organization and exocytosis have emerged as central pathological mechanisms in PD, involving dysregulation of αSyn, SNARE proteins, SV pools and composition, and kinase signaling, as discussed below.
α-Synuclein (PARK1/PARK4/SNCA): αSyn is a small presynaptic protein that exists in equilibrium between an intrinsically disordered monomer in the cytoplasm and an α-helical multimer on SVs. This equilibrium is modulated by SV lipids, protein:lipid ratio, proteins and their PTMs, some of which are altered in PD.46 SV binding of αSyn is considered to be neuroprotective under physiological conditions.47–52 However, aging and expression of human αSyn alter SV lipid composition and protein:lipid ratio,8 potentially compromising these protective mechanisms. SV-bound αSyn clusters SVs and chaperones SNARE-complex assembly to maintain neuronal firing.53 These activities are impaired by αSyn mutants that reduce its affinity towards SVs, such as PD-linked A30P,49,54 and with excess αSyn by interfering with synapsin's ability to form condensates.55 Oligomeric WT, and PD-linked A30P and A53T αSyn retain the clustering ability but inhibit SNARE-mediated liposome fusion in vitro.56,57 E46K oligomers perforate VAMP2-containing vesicles and reduce membrane lipid mobility in vitro.57 αSyn has also been shown to tubulate liposomes in a SNARE-dependent manner, with enhanced effects for A30P and E46K variants and elevated αSyn levels, resulting in inhibition of SNARE-mediated membrane fusion.58 Similarly, αSyn PFFs disrupt SV lipid bilayers in vitro.59
αSyn pathology combines loss-of-function and gain-of-toxic function effects and is linked to synapse loss.43 Dose of and mutations in αSyn drive early-onset PD where αSyn aggregates to form Lewy bodies and Lewy neurites.46 Recent studies have highlighted the prodromal effects of αSyn. Rats overexpressing A53T αSyn reveal dysregulation of synaptic proteins such as β-synuclein and RIMBP2, and undergo dopaminergic neurodegeneration.60 αSyn PFFs injected into the rat striatum reduce EPSCs and increase substantia nigra dopamine neuron firing.61 In mice overexpressing human αSyn, dopamine release shows age-dependent dysfunction, with enhancements in 3 month-old animals preceding behavioral changes and a reduction in 12 month-old symptomatic mice,62 highlighting an early stage of compensation followed by a later stage of dopamine release failure. Oligomeric αSyn reduces excitatory neurotransmission and VGLUT1 expression at striatal glutamatergic terminals.63 Extracellular monomeric αSyn disrupts neuronal firing through lowering cholesterol in presynaptic membranes, increasing tonic but reducing depolarization-evoked SV recycling and glutamate release.64 Cultured rat glutamatergic neurons expressing human αSyn at familial triplication levels maintain presynaptic proteins and synaptic transmission and plasticity,65 while iPSC-derived human cortical glutamatergic neurons with αSyn gene triplication reveal decreased spontaneous and evoked SV trafficking,66 suggesting that species- or cell-type-specific differences may influence the manifestation of αSyn-induced synaptic dysfunction.
VAMP2/synaptobrevin-2 (VAMP2/SYB2) is the R-SNARE located on SVs that together with syntaxin-1 and SNAP-25 forms the presynaptic SNARE complex, a stable four-helix bundle enabling SV exocytosis.5 VAMP2 and αSyn function are tightly linked. Their interaction modulates both SV clustering and SNARE complex assembly,53 and VAMP2 reduces αSyn aggregation in vitro.67 In PD brain tissue, SV2A and VAMP2 levels are elevated in early disease stages but reduced in late stages,68 establishing VAMP2 as a stage-dependent regulator of synaptic dysfunction. Supportively, VAMP2 levels are reduced in PD patient extracellular vesicles, providing an accessible biomarker for disease.69 This reduction reflects broader disease-stage-dependent changes in VAMP2 levels: in mice, overexpressing human αSyn in the dorsal raphe nucleus results in a depressive phenotype with synaptic pathology, lower MAP2 and PSD95 levels, and elevated levels of SV2A and VAMP2,68 indicating possible compensatory mechanism for early synaptic loss to maintain neurotransmission and dopamine homeostasis. Yet, this compensation is not sustainable, resulting in reduced SV2A and VAMP2 levels and synaptic failure with αSyn accumulation and Lewy body formation in later disease stages.
SV2: SV2C is a SV transmembrane glycoprotein that is enriched in dopaminergic neurons70 where it contributes to dopamine homeostasis and assists in retaining neurotransmitters within SVs. SV2C is a risk locus for PD,71–73 and its expression pattern is disrupted in PD brains.74 It is transcriptionally downregulated in PD substantia nigra, and PET scans reveal reduced levels of SV2A that correlate with symptom severity and disease duration, suggesting progressive synaptic loss in PD.75 Decreased levels of SV2C have also been reported in serum of PD patients.76 SV2C binds synaptotagmin-1,77 and enhances uptake and retention of dopamine in vesicular compartments of HEK293 cells and dopamine retention in isolated SVs from mouse brains.78 SV2C also contributes to MPTP resistance in mice,78 and has a protective role in PD that is associated with cigarette smoking.79 In flies expressing human αSyn, nicotine rescues their PD phenotype, mediated via two SV2 fly orthologues.80
LRRK2 (PARK8) is a cytoplasmic protein with GTPase and kinase activity81 that phosphorylates rab proteins to regulate SV trafficking. Its N-terminus mediates interactions with synapsin I, α-tubulin and β-actin.82 LRRK2 variants linked to late-onset autosomal dominant PD83 increase its kinase activity, leading to aberrant protein phosphorylation and dysregulation of SV protein trafficking.84 Overexpression of N-terminally truncated or E193K LRRK2 diminishes binding to SVs in vitro and enhances SV fusion.82 iPSC-derived human glutamatergic neurons carrying LRRK2 variants demonstrate hyperphosphorylated rab3a and impaired interaction with the motor adapter protein MADD, leading to reduced anterograde transport of SV precursors and mislocalization of synaptic proteins such as synaptophysin and VAMP2.85 At the synapse, pathogenic LRRK2 variants impair active zone organization and produce circuit-specific synaptic dysfunction that precedes motor symptoms. In LRRK2 mutant mice, increased rab3a/c phosphorylation in the substantia nigra was shown to reduce rab3 binding to active zone organizers RIM1 and RIM2, disrupting active zone structure and resulting in decreased evoked dopamine release in vivo.86 Human-derived LRRK2G2019S neurons show reduced frequency of spontaneous excitatory synaptic currents.87 In LRRK2G2019S mice, glutamatergic transmission is impaired across multiple basal ganglia circuits months before motor symptom onset, with dopamine neurons displaying reduced presynaptic VGLUT1 and diminished glutamate release probability onto substantia nigra neurons.88 Notably, this dysfunction is not uniform: substantia nigra pars reticulata GABAergic neurons display increased paired-pulse depression of glutamatergic currents, indicating elevated glutamate release probability,89 demonstrating that circuit dysfunction operates through bidirectional mechanisms rather than simple suppression. These findings establish LRRK2 as a critical player linking molecular defects to presynaptic dysfunction in PD.
Synaptic vesicle endocytosis
SV endocytosis represents a critical homeostatic mechanism that recycles vesicular membranes and proteins after exocytosis to replenish releasable SV pools. Its disruption constitutes a central pathogenic mechanism in PD, with multiple disease-linked proteins converging on this pathway. As discussed below, SV protein and lipid dyshomeostasis drive specific changes in endocytosis that trigger synaptic dysfunction and PD pathology.
LRRK2 (PARK8) phosphorylates multiple proteins involved in SV endocytosis, including NSF, auxilin, endophilin A1, synaptojanin-1, dynamin, and AP2.84,90 LRRK2 knockout dysregulates AP2 function in cultured neurons, leading to impaired clathrin-mediated endocytosis.91 Transcriptomic and proteomic analysis of iPSC-derived dopaminergic neurons expressing mutated LRRK2 reveal impairments in SV endocytosis, which has been reproduced in transgenic rat brain tissue,92 along with increased numbers of clathrin- and endophilin-positive puncta in PD brains.92 Untargeted lipidomic analysis reveals altered glycerolipid and sphingolipid metabolism in PD patients carrying LRRK2 variants.93
αSyn (PARK1/PARK4/SNCA): Several studies highlight αSyn's role in clathrin-mediated endocytosis (CME). It colocalizes with PI(4,5)P2 and phosphorylated AP2 in clathrin-coated pits and enhances CME by increasing plasma membrane PI(4,5)P2 levels. PD-linked A30P, E46K and A53T αSyn further upregulate CME.94 These effects are mediated by synaptojanin-1: WT or A53T αSyn overexpression reduces synaptojanin-1 levels, whereas reintroduction of synaptojanin-1 rescues CME defects by restoring PI(4,5)P2 homeostasis.95 PI(4,5)P2-bound αSyn also increases clathrin lattice size and vesicle curvature via indirect interactions with AP180, which together with AP2 recruits clathrin to SV membranes.96 In lamprey giant neurons, high αSyn levels prevent clathrin uncoating during CME by Hsc70 sequestration, leading to SV depletion.97
Synaptojanin-1 (PARK20/SYNJ1) is a brain-specific inositol phosphatase involved in SV uncoating during CME98 via dephosphorylation of PI(4,5)P2 to PI99 and removal of clathrin adapters from the SV membrane. Its C-terminus is phosphorylated by LRRK2 and interacts with SH3 domain proteins including endophilin A1 and amphiphysin,100 positioning it as a central node in the endocytic machinery. Synaptojanin-1 is also a substrate for parkin ubiquitination,101 linking it to the PINK1/parkin pathway. SYNJ1 mutations cause partial loss of function, leading to autosomal recessive juvenile or early-onset parkinsonism with variable presentations including seizures, cognitive impairment and atypical symptoms, or typical PD.102 Various SYNJ1 variants alter its transcript levels, phosphatase activity, binding to AP2102 and/or sphingolipid, inositol, and inositol phosphate metabolism.103 Mutations within its Sac1 domain alter activity-dependent endocytosis.104 Synaptojanin-1 transcript levels are lower in PD patient blood and brain tissue,105 accompanied by PI(3,4,5)P3 accumulation and colocalization with phosphorylated αSyn.106 C-terminal cleavage in PD brains leads to reduced phosphatase activity and lack of binding to endophilin A1, amphiphysin or dynamin-1.107 Furthermore, synaptojanin-1 dysfunction triggers a cascade of presynaptic pathology: C-terminal cleavage causes presynaptic dysfunction, motor deficits, and degeneration of dopaminergic neurons in A53T αSyn-overexpressing mice107; synaptojanin-1 knockout in SH-SY5Y cells increases αSyn and PI(3,4,5)P3 levels106; and treating primary neurons with PI(3,4,5)P3 or PTEN PI(3,4,5)P3 phosphatase inhibitor causes αSyn aggregation, possibly via binding to PI(3,4,5)P3.106 Similarly, elevated levels of PI(4,5)P2 disrupt SV endocytosis in primary neurons.108 In C. elegans, mutant synaptojanin-1 produces αSyn aggregation and motor dysfunction.106 In mice, synaptojanin-1 heterozygosity results in PD-like pathology with motor dysfunction, phosphorylated αSyn accumulation, impaired autophagy and elevated neuronal PI(4,5)P2 levels.108
Endophilin A1 (SH3GL2): SH3GL2 is a risk gene for PD.109 Endophilin A1 interacts with dynamin, synaptojanin-1, VGLUT1 and other synaptic proteins, and is ubiquitinated by parkin110,111 and phosphorylated by LRRK2.112 Membrane recruitment of dynamin-1 and synaptojanin-1 by endophilin A1 is crucial for ultrafast SV endocytosis, and for removing clathrin from SVs that bud off from endosomes to regenerate SVs.113,114 Lack of endophilins A1 and A2 or synaptojanin-1 prevents formation of the neck at endocytic pits,113 impairing endocytosis.
Synaptotagmin-11 (SYT11) is ubiquitinated by parkin,115 inhibits SV endocytosis through interaction with endophilin A1,116 and inhibits spontaneous neurotransmission.117 Single nucleotide polymorphisms in SYT11 are associated with PD.118 Synaptotagmin-11 deficiency enhanced SV endocytosis in primary neurons.116 In dopaminergic circuits, synaptotagmin-11 plays a complex, context-dependent role that varies with circuit state. Synaptotagmin-11 upregulation during early postnatal development in parkin knockout mice serves as a compensatory mechanism that conceals dopaminergic release deficits. However, synaptotagmin-11 overexpression or accumulation in adult dopaminergic neurons of the substantia nigra impairs dopamine release and induces motor and depressive-like behaviors.119
Parkin (PARK2) is an E3 ubiquitin ligase120 that is associated with sporadic PD.121 Parkin variants or multiplications lead to autosomal recessive juvenile122 or early-onset PD.121 Parkin is activated by PINK1,123 another gene linked to PD, but can also be independently activated via phosphorylation by CamKII for its recruitment to SVs.101 Parkin regulates SV endocytosis through ubiquitination of endophilin A1,111 synaptojanin-1101 and synaptotagmin-11.115 Parkin mutations impair these mechanisms, causing reduced SV numbers, enlarged SV diameter, dopamine dysfunction, and trafficking deficits in patient-derived dopaminergic neurons, which can be reversed by expression of endophilin A1.101 PINK1/parkin mutations produce early, neuron-type-specific presynaptic changes that reflect distinct circuit compensation. In PINK1 knockout rats, glutamate release onto dorsal striatal neurons and spontaneous EPSC activity are increased, but SV replenishment is impaired.124 Patient-derived dopaminergic neurons carrying both PINK1 and parkin mutations show heightened synaptic activity, while their hippocampal neurons display reduced synaptic function, highlighting cell-type differences in response.125
Auxilin (PARK19/DNAJC6) is a neuronal cofactor for the Hsc70 ATPase that is phosphorylated by LRRK2.90 It binds phosphatidylinositol and clathrin, removing clathrin from adapter proteins during CME via triggering conformational changes, and prevents disassembly of spontaneously assembled clathrin cages.126 Auxilin splice variants, single site mutants and truncations lead to partial or complete loss of function and cause autosomal recessive juvenile or early-onset parkinsonism with seizures and cognitive impairment that is variably L-DOPA responsive.127 In PD patients, nonsense mutations trigger reduced auxilin levels in CSF and fibroblasts, elevated CSF levels of GAK/auxilin-2 (a compensatory family member), and decreased CSF levels of tyrosine hydroxylase, DAT and VMAT, indicating dopamine dyshomeostasis which was confirmed by reduced DAT availability in PET imaging.128 Patient-derived dopaminergic neurons carrying auxilin mutations demonstrate auxilin deficiency, leading to reduced SV numbers due to aberrant SV trafficking and CME, with bulk RNA sequencing revealing downregulation of genes encoding the presynaptic proteins VGLUT1, VGLUT2, and VMAT2.129 Auxilin dysfunction triggers widespread presynaptic pathology: auxilin knockout mice develop PD-like motor deficits rescued by L-DOPA, and demonstrate SV sorting defects, empty clathrin cages, dopamine accumulation and oxidation in the cytoplasm, increased autophagy, and trapped DAT, decreasing dopamine reuptake from the synaptic cleft.130 AuxilinR927G mice were shown to have a PD-like phenotype and reveal reduced SV numbers, decreased interactions of auxilin with clathrin and Hsc70, and clathrin uncoating defects alongside increased clathrin-coated vesicles.131 In the fly, auxilin mutant variants disrupt the J domain, causing synaptic dysfunction, age-dependent decrease of auxilin levels, motor deficits, seizures, lipid dyshomeostasis and neurodegeneration, which can be rescued by overexpression of synaptojanin-1.132 Interestingly, loss of function of auxilin and synaptojanin-1 produces synergistic effects on clathrin uncoating, with increased vulnerability of nigrostriatal neurons and significantly shortened mouse lifespan, with double mutant mice accumulating not only clathrin-coated vesicles but also SNAP25 and SV2C.126 Auxilin knockout mice demonstrate impaired presynaptic plasticity and altered sensory processing, emphasizing the vital role of endocytic machinery components in synaptic maintenance.133
Cyclin G associated kinase (Auxilin 2, GAK/DNAJC26) is a ubiquitously expressed J-domain protein and a paralog of auxilin which can compensate for auxilin dysfunction. Similarly to auxilin, it chaperones uncoating of clathrin cages via binding to Hsc70.134 It also modulates Na+/K+ -ATPase trafficking to the plasma membrane.135 GAK gene polymorphism increases PD risk and is associated with increased αSyn levels.136 GWAS analysis revealed altered GAK transcript levels that are linked to expression of synapse-associated genes.137 In cultured neurons, GAK inhibition results in reduced neurite outgrowth and synapse formation.138
SCAMP5 is a SV protein that interacts with the SV protein synaptotagmin-1139 the vesicular Na+(K+)/H+ exchanger NHE6 to recruit NHE6 to glutamatergic terminals,140 and the adapter protein AP2141 to modulate clearance of active zone release sites and SV endocytosis. While traditionally linked to autism,142 seizures and developmental delay,143 recently, R91W SCAMP5 was found to cause autosomal recessive juvenile PD with seizures.139,144 SCAMP5 deficiency triggers multiple pathological mechanisms: SCAMP5 R91W disrupts its localization and interactions with synaptotagmin-1 in mice139; SCAMP5 deficiency in PC12 cells leads to elevated αSyn levels, αSyn oligomerization, deficient dopamine secretion, and apoptosis144; and SCAMP5 knockdown in SH-SY5Y cells results in lowered secretion of αSyn-containing exosomes, which is rescued by expression of wild type but not mutant SCAMP5.144 Further, SCAMP5 knockdown inhibits recruitment of NHE6 to glutamatergic terminals, causing SV hyperacidification and decreased resting glutamate release in rat primary neurons.145 This specific effect on glutamatergic neurons reflects its preferential association with VGLUT2-SVs compared to VMAT2-SVs in dopaminergic neurons co-expressing both transporters, suggesting specific involvement of VGLUT2 recycling in PD pathology.146 In zebrafish, knockout of scamp5a causes PD-like motor phenotypes, degeneration of dopaminergic neurons and dopamine dyshomeostasis, with transcriptome analysis showing upregulated JNK signaling associated with cell apoptosis.144 SCAMP5R91W knockin mice develop sound-sensitive juvenile epilepsy.139
Sac2 (INPP5F) is a PI-4-P phosphatase that interacts with rab5 to be recruited to endosomes.147 It is also a PD risk gene.148 Knockout in mice does not produce a phenotype on its own, but exaggerates phenotypes of mutated synaptojanin-1, causing earlier lethality, elevated parkin levels, and accumulation of amphiphysin-2, clathrin light chain, auxilin and synaptojanin-1.149
DJ-1 (PARK7) is enriched in presynaptic terminals, where it stimulates VMAT2 activity,150,151 interacts with DAT to increase dopamine reuptake from the synaptic cleft,152 protects from oxidative stress via neutralizing reactive oxygen species,153,154 and reduces αSyn aggregation in vitro.155 Mutations in DJ-1 lead to autosomal recessive early-onset PD.156 DJ-1 knockout neurons reveal reduced SV endocytosis and re-availability, which is rescued by wild type but not mutant DJ-1.157 Furthermore, cultured mouse knockout neurons reveal slower closing of the exocytotic fusion pore, leading to prolonged exocytosis.155 Knockout in iPSC-derived human dopaminergic neurons results in reduced VMAT2 levels and activity, which leads to impaired dopamine uptake into SVs and cytoplasmic dopamine oxidation.151 Additionally, DJ-1 deficiency causes aberrant SV morphology including enlarged SVs, membrane elongation/tubulation, and accumulation of clathrin, suggesting disrupted SV uncoating and trafficking.151 In mice, loss of DJ-1 triggers widespread presynaptic dysfunction, reduced activity of the postsynaptic D2 dopamine receptor, and selective neurodegeneration.158 In rats, DJ-1 loss produces motor impairments, degeneration of dopaminergic neurons, elevated tyrosine hydroxylase and increased serotonin transporter availability in cortical tissue shown via ligand-binding autoradiography, suggesting dopamine and serotonin dyshomeostasis.159
Glucocerebrosidase (GBA1) breaks down glucosylceramide to glucose and ceramide in lysosomes. Its absence causes substrate accumulation. Loss of function mutations in GBA1 lead to Gaucher's disease, an autosomal recessive lysosomal storage disorder, and to an increased risk for PD160 and dementia with Lewy bodies,161 even when heterozygous. Reduced GBA activity is also observed in sporadic PD.162 GBA and αSyn function are intertwined. GBAL444P mice reveal cognitive decline without motor phenotypes or αSyn pathology, while αSynA30P mice show motor deficits and αSyn pathology without cognitive decline. Mice carrying both mutants combine and exacerbate these phenotypes. Interestingly, GBA mutant mice reveal significant dysregulation of genes and proteins involved in SV trafficking and synapse assembly, including rab26, Hsc70, parkin, endophilin A1, PI(4,5)P2, along with reduced SV numbers and clathrin-coated vesicles.163 Supportively, GBA variants produce distinct presynaptic effects, depending on the mutation: GBAE326K neurons display reductions in both amplitude and rate of EPSCs,164 while GBAN370S neurons exhibit increased spontaneous firing and stronger evoked dopamine release,165 suggesting variant-specific mechanisms in PD pathogenesis.
Presynaptic αSYN pathology triggers postsynaptic and curcuit-level dysfunction
Above, we have highlighted how dysfunctional SV-related pathways drive physiological changes underlying PD. This presynaptic pathology initiates postsynaptic changes and circuit reorganization across multiple brain regions and in multiple neuron types. Here, we briefly outline how αSyn pathology perturbs these processes, emphasizing that presynaptic dysfunction does not occur in isolation but is tightly coupled with downstream synaptic and circuit-level alterations.
In αSynA53T overexpressing mice, early dopamine loss impairs synaptic plasticity and dendritic spine remodeling.166 αSyn PFF injections into mice block long-term synaptic plasticity, which is reversible by L-DOPA administration.61 Regional vulnerability differs markedly. Overexpression of αSyn in the dorsal motor nucleus (DMV) causes a 57% reduction in the firing rate via Kv4 channelopathy167 which drives constipation. Phosphorylated αSyn in DMV neurons is found already in 2-month-old BAC-SNCA mice, possibly underlying early behavioral alterations.168 αSyn pathology in the locus coeruleus (LC) triggers calcium-activated potassium channel dysfunction causing neuronal hyperexcitability169 that drives noradrenaline/serotonin system dysregulation, widespread propagation to the hippocampus/prefrontal cortex/dorsal raphe, and prodromal cognitive and anxiety phenotypes170 – all occurring before LC neurodegeneration.171 In midbrain regions, αSyn accumulation produces differential circuit effects: substantia nigra pars compacta (SNc) dopamine neurons show increased baseline firing and impaired homeostatic regulation, while ventral tegmental area (VTA) neurons remain relatively unaffected,172 establishing circuit-specific vulnerability that may explain the selective SNc neurodegeneration in PD. Across cortical, limbic, and sensory regions, αSyn pathology produces region-specific changes: anterior cingulate cortex shows increased excitatory transmission,173 prefrontal cortex displays reduced GDNF and dendritic spine loss,174 hippocampus loses GluA1 AMPA receptors and synaptic plasticity,175 basolateral amygdala selectively loses cortical glutamate transmission,176 accompanied by structural alterations in pre- and postsynaptic terminals,177 while the olfactory bulb shows impaired GABA transmission.178 αSyn-induced presynaptic dysfunction triggers also secondary neurotransmitter imbalance. Downregulation of GABA reuptake transporters elevates tonic GABA inhibition of dopamine neurons, particularly in the dorsolateral striatum.179 Brain imaging of wild type mice overexpressing human αSyn in raphe serotonin neurons reveals a hypo-connectivity in the cortico-limbic-raphe network and development of a depressive phenotype, consequences of progressive loss of serotonin transporter-positive projections and impaired pre-synaptic vesicle trafficking in serotonergic terminals.68 These findings establish hierarchical presynaptic αSyn pathology that drives postsynaptic plasticity impairments and circuit imbalances which progressively spread through interconnected pathways, providing an explanation for the varied symptoms in PD.
Conclusions
Recent findings have established a paradigm shift in PD where SV dysfunction is not just an early event in disease but a central, targetable mechanism that drives neurodegeneration (Table 1, Figure 1). Impaired synaptic transmission occurs across both genetic and sporadic forms of PD, and triggers cascading dysfunction throughout brain circuits. Synaptic deficits appear months before motor symptoms and neuronal death, creating a therapeutic window for early intervention that should be explored in future studies. While targeting SV dysfunction is a viable therapeutic strategy, very little has been attempted. Synaptic dysfunction is not only associated with PD but also with Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis and frontotemporal dementia.180 Drugs tested for promoting and rebalancing synaptic function in these diseases have been traditionally used for psychiatric conditions and epilepsy, and target receptors to promote synapse function and enhance synaptic strength, block postsynaptic receptors, focus on signaling pathways to prevent axon degeneration, and target the inhibitory/excitatory balance.181 Approaches specific to SV proteins are rare. Levetiracetam targets the vesicle protein SV2A to reduce hippocampal hyperactivity in mild cognitive impairment182,183 and improved executive function and spatial memory among participants, but did not improve a composite cognitive function score.184 Brivaracetam, a more selective, higher-affinity SV2A ligand with rapid brain penetration, shows a favorable cognitive profile in epilepsy patients and in some studies is associated with stabilization or modest improvement of attention and executive function.185,186 These SV2A ligands demonstrate that pharmacological targeting of SV proteins is feasible in humans, but they remain isolated examples. Systematic discovery and testing of SV-focused therapeutics for neurodegenerative diseases require much greater attention and should be explored to maintain synapse health and/or repair synapses before the damage is irreversible.
Footnotes
Ethical considerations
Not applicable
Consent to participate
Not applicable
Consent for publication
N/A.
Author contributions
J.C. and V.I. researched the literature and wrote the initial draft of the manuscript. All authors worked together to refine and revise the manuscript and generate the figure and table.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the American Parkinson Disease Foundation (J.C.) and from NIH (R01NS113960, R01NS121077, R01NS126342 and R01NS136423 to J.B).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
N/A.
ORCID iDs
Julita Chlebowicz
Violetta Ivanova
Jacqueline Burré
References
1.
GaoVCrawfordCVBurréJ. The gut-brain axis in Parkinson's disease. Cold Spring Harb Perspect Med2025; 15: 20250107.
2.
HenrichMTOertelWHSurmeierDJ, et al.Mitochondrial dysfunction in Parkinson's disease - a key disease hallmark with therapeutic potential. Mol Neurodegener2023; 18: 83. 20231111.
3.
MachtelRBorosFADobertJP, et al.From lysosomal storage disorders to Parkinson's disease - challenges and opportunities. J Mol Biol2023; 435: 167932. 20221223.
4.
NgXYCaoM. Dysfunction of synaptic endocytic trafficking in Parkinson's disease. Neural Regen Res2024; 19: 2649–2660. 20240301.
TakamoriSHoltMSteniusK, et al.Molecular anatomy of a trafficking organelle. Cell2006; 127: 831–846.
7.
TaoufiqZNinovMVillar-BrionesA, et al.Hidden proteome of synaptic vesicles in the mammalian brain. Proc Natl Acad Sci U S A2020; 117: 33586–33596. 20201221.
8.
GaoVChlebowiczJGaskinK, et al.Synaptic vesicle-omics in mice captures signatures of aging and synucleinopathy. Nat Commun2025; 16: 4079. 20250501.
9.
BolzSHauckeV. Biogenesis and reformation of synaptic vesicles. J Physiol2025; 603: 5965–5979. 20241005.
10.
KulkarniASBurnsMRBrundinP, et al.Linking alpha-synuclein-induced synaptopathy and neural network dysfunction in early Parkinson's disease. Brain Commun2022; 4: fcac165. 20220622.
11.
YoshinoHTomiyamaHTachibanaN, et al.Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology2010; 75: 1356–1361.
12.
KrebsCEKarkheiranSPowellJC, et al.The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat2013; 34: 1200–1207. 20130719.
13.
QuadriMFangMPicilloM, et al.Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat2013; 34: 1208–1215. 20130806.
14.
BlakelyRDEdwardsRH. Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harb Perspect Biol2012; 4: 20120201.
15.
ZhouQPetersenCCNicollRA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol2000; 525: 195–206.
16.
AbbasYMWuDBuelerSA, et al.Structure of V-ATPase from the mammalian brain. Science2020; 367: 1240–1246.
17.
MaLOuyangQWerthmannGC, et al.Live-cell microscopy and fluorescence-based measurement of luminal pH in intracellular organelles. Front Cell Dev Biol2017; 5: 71. 20170821.
18.
WangCJiangWLeitzJ, et al.Structure and topography of the synaptic V-ATPase-synaptophysin complex. Nature2024; 631: 899–904. 20240605.
19.
KravcenkoURuwoltMKrollJ, et al.Molecular architecture of synaptic vesicles. Proc Natl Acad Sci U S A2024; 121: e2407375121. 20241127.
20.
WangRLongTHassanA, et al.Cryo-EM structures of intact V-ATPase from bovine brain. Nat Commun2020; 11: 3921. 20200806.
21.
MillerGWEricksonJDPerezJT, et al.Immunochemical analysis of vesicular monoamine transporter (VMAT2) protein in Parkinson's disease. Exp Neurol1999; 156: 138–148.
22.
PiflCRajputAReitherH, et al.Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J Neurosci2014; 34: 8210–8218.
23.
BeauchampLCVillemagneVLFinkelsteinDI, et al.Reduced striatal vesicular monoamine transporter 2 in REM sleep behavior disorder: imaging prodromal parkinsonism. Sci Rep2020; 10: 17631. 20201023.
24.
ValliMChoSSUribeC, et al.VMAT2 Availability in Parkinson's disease with probable REM sleep behaviour disorder. Mol Brain2021; 14: 165. 20211110.
25.
RilstoneJJAlkhaterRAMinassianBA. Brain dopamine-serotonin vesicular transport disease and its treatment. N Engl J Med2013; 368: 543–550. 20130130.
26.
GlattCEWahnerADWhiteDJ, et al.Gain-of-function haplotypes in the vesicular monoamine transporter promoter are protective for Parkinson disease in women. Hum Mol Genet2006; 15: 299–305. 20051208.
27.
SveinsdottirHSDeckerAChristensenC, et al.Motility phenotype in a zebrafish vmat2 mutant. PLoS One2022; 17: e0259753. 20220105.
28.
BucherMLBarrettCWMoonCJ, et al.Acquired dysregulation of dopamine homeostasis reproduces features of Parkinson's disease. NPJ Parkinsons Dis2020; 6: 34. 20201113.
29.
Gonzalez-SepulvedaMCompteJCuadrosT, et al.In vivo reduction of age-dependent neuromelanin accumulation mitigates features of Parkinson's disease. Brain2023; 146: 1040–1052.
30.
LeeHKimHJMinJS, et al.HDAC4/5 Inhibitor, LMK-235 improves animal voluntary movement in MPTP-induced Parkinson's disease model. Pharmacol Res Perspect2025; 13: e70057.
31.
El-HabtaRAf BjerkenSVirelA. N-acetylcysteine increases dopamine release and prevents the deleterious effects of 6-OHDA on the expression of VMAT2, alpha-synuclein, and tyrosine hydroxylase. Neurol Res2024; 46: 406–415. 20240318.
32.
ZhaoMZhouYShengR, et al.Gastrodin relieves Parkinson's disease-related motor deficits by facilitating the MEK-dependent VMAT2 to maintain dopamine homeostasis. Phytomedicine2024; 132: 155819. 20240611.
33.
ConradWSOriolLKollmanGJ, et al.Proportion and distribution of neurotransmitter-defined cell types in the ventral tegmental area and substantia nigra pars compacta. Addict Neurosci2024; 13: 20241028.
34.
FujiseKMishraJRosenfeldMS, et al.Synaptic vesicle characterization of iPSC-derived dopaminergic neurons provides insight into distinct secretory vesicle pools. NPJ Parkinsons Dis2025; 11: 16. 20250109.
35.
FremeauRTJrTroyerMDPahnerI, et al.The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron2001; 31: 247–260.
36.
HerzogEBellenchiGCGrasC, et al.The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci2001; 21: RC181.
37.
VaroquiHSchaferMKZhuH, et al.Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci2002; 22: 142–155.
38.
KochmanskiJKuhnNCBernsteinAI. Parkinson's disease-associated, sex-specific changes in DNA methylation at PARK7 (DJ-1), SLC17A6 (VGLUT2), PTPRN2 (IA-2beta), and NR4A2 (NURR1) in cortical neurons. NPJ Parkinsons Dis2022; 8: 120. 20220923.
39.
SteinkellnerTConradWSKovacsI, et al.Dopamine neurons exhibit emergent glutamatergic identity in Parkinson's disease. Brain2022; 145: 879–886.
40.
BuckSADe MirandaBRLoganRW, et al.VGLUT2 Is a determinant of dopamine neuron resilience in a rotenone model of dopamine neurodegeneration. J Neurosci2021; 41: 4937–4947. 20210423.
41.
MooreCHelmsMLNipperMA, et al.Dopamine loss alters glutamate synapses and transporters in the medial prefrontal cortex and anxiety-related behaviour in a male MPTP rodent model of Parkinson's disease. Eur J Neurosci2024; 60: 6195–6215. 20241021.
42.
ClarkKAWhiteAJPaslawskiW, et al.Parkinson disease-associated toxic exposures selectively up-regulate vesicular glutamate transporter vGlut2 in a model of human cortical neurons. Mol Biol Cell2025; 36: br4. 20250102.
43.
SahSSauerbeckADGuptaJ, et al.Progressive vulnerability of cortical synapses in alpha-synucleinopathy. bioRxiv2025: 1–41. doi:10.1101/2024.06.20.599774
44.
LuoYQiaoLLiM, et al.Global, regional, national epidemiology and trends of Parkinson's disease from 1990 to 2021: findings from the global burden of disease study 2021. Front Aging Neurosci2024; 16: 1498756. 20250110.
45.
BuckSASteinkellnerTAslanoglouD, et al.Vesicular glutamate transporter modulates sex differences in dopamine neuron vulnerability to age-related neurodegeneration. Aging Cell2021; 20: e13365. 20210428.
46.
BurréJSharmaMSudhofTC. Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med2018; 8: 20180301.
47.
NarayananVScarlataS. Membrane binding and self-association of alpha-synucleins. Biochemistry2001; 40: 9927–9934.
YooGYeouSSonJB, et al.Cooperative inhibition of SNARE-mediated vesicle fusion by alpha-synuclein monomers and oligomers. Sci Rep2021; 11: 10955. 20210526.
57.
YooGAnHJYeouS, et al.alpha-Synuclein disrupts vesicle fusion by two mutant-specific mechanisms. Mol Cells2022; 45: 806–819. 20221111.
58.
LiQHuTLiF, et al.alpha-Synuclein drives SNARE-dependent tubular remodeling of vesicles. J Am Chem Soc2025; 147: 29829–29837. 20250806.
59.
StephensADVillegasAFChungCW, et al.Alpha-Synuclein fibril and synaptic vesicle interactions lead to vesicle destruction and increased lipid-associated fibril uptake into iPSC-derived neurons. Commun Biol2023; 6: 526. 20230515.
60.
Belloso-IguerateguiAZamarbideMMerino-GalanL, et al.Hippocampal synaptic failure is an early event in experimental parkinsonism with subtle cognitive deficit. Brain2023; 146: 4949–4963.
61.
TozziASciaccalugaMLoffredoV, et al.Dopamine-dependent early synaptic and motor dysfunctions induced by alpha-synuclein in the nigrostriatal circuit. Brain2021; 144: 3477–3491.
62.
Medina-LuqueJPiechocinskiPFeyenP, et al.Striatal dopamine neurotransmission is altered in age- and region-specific manner in a Parkinson's disease transgenic mouse. Sci Rep2024; 14: 164. 20240102.
63.
BellingacciLSciaccalugaMMegaroA, et al.Oligomeric alpha-synuclein causes early synaptic dysfunction of the corticostriatal pathway associated with non-motor symptoms. NPJ Parkinsons Dis2025; 11: 220. 20250729.
64.
LazarevicVYangYPaslawskiW, et al.alpha-Synuclein induced cholesterol lowering increases tonic and reduces depolarization-evoked synaptic vesicle recycling and glutamate release. NPJ Parkinsons Dis2022; 8: 71. 20220607.
65.
SantosPIGarcia-PlazaIHShaibA, et al.Glutamatergic synaptic resilience to overexpressed human alpha-synuclein. NPJ Parkinsons Dis2025; 11: 238. 20250812.
WangCZhangKCaiB, et al.VAMP2 Chaperones alpha-synuclein in synaptic vesicle co-condensates. Nat Cell Biol2024; 26: 1287–1295. 20240701.
68.
Miquel-RioLJerico-EscolarJSarries-SerranoU, et al.Early synaptic changes and reduced brain connectivity in PD-like mice with depressive phenotype. NPJ Parkinsons Dis2025; 11: 242. 20250813.
69.
AgliardiCMeloniMGueriniFR, et al.Oligomeric alpha-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson's disease. Neurobiol Dis2021; 148: 105185. 20201118.
70.
DunnARHoffmanCAStoutKA, et al.Immunochemical analysis of the expression of SV2C in mouse, macaque and human brain. Brain Res2019; 1702: 85–95. 20171221.
71.
FooJNChewEGYChungSJ, et al.Identification of risk loci for Parkinson disease in asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol2020; 77: 746–754.
72.
GroverSKumar-SreelathaAABobbiliDR, et al.Replication of a novel Parkinson's locus in a European ancestry population. Mov Disord2021; 36: 1689–1695. 20210324.
73.
TanZLinYZhouM, et al.Correlation of SV2C rs1423099 single nucleotide polymorphism with sporadic Parkinson's disease in Han population in southern China. Neurosci Lett2023; 813: 137426. 20230805.
74.
DunnARStoutKAOzawaM, et al.Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci U S A2017; 114: E2253–E2262. 20170228.
75.
HolmesSEHonharPTinazS, et al.Synaptic loss and its association with symptom severity in Parkinson's disease. NPJ Parkinsons Dis2024; 10: 42. 20240224.
76.
WuJWuWJiangP, et al.Identification of SV2C and DENR as key biomarkers for Parkinson's disease based on bioinformatics, machine learning, and experimental verification. J Mol Neurosci2024; 74: 6. 20240108.
77.
SchivellAEMochidaSKensel-HammesP, et al.SV2A And SV2C contain a unique synaptotagmin-binding site. Mol Cell Neurosci2005; 29: 56–64.
78.
BucherMLDunnARBradnerJM, et al.Synaptic vesicle glycoprotein 2C enhances vesicular storage of dopamine and counters dopaminergic toxicity. Eur J Neurosci2024; 59: 2483–2501. 20240326.
79.
Hill-BurnsEMSinghNGangulyP, et al.A genetic basis for the variable effect of smoking/nicotine on Parkinson's disease. Pharmacogenomics J2013; 13: 530–537. 20121002.
80.
OlsenALClemensSGFeanyMB. Nicotine-Mediated rescue of alpha-synuclein toxicity requires synaptic vesicle glycoprotein 2 in Drosophila. Mov Disord2023; 38: 244–255. 20221123.
81.
PischeddaFPiccoliG. LRRK2 At the pre-synaptic site: a 16-years perspective. J Neurochem2021; 157: 297–311. 20210205.
82.
MarkuACarrionMDPPischeddaF, et al.The LRRK2 N-terminal domain influences vesicle trafficking: impact of the E193 K variant. Sci Rep2020; 10: 3799. 20200302.
83.
MataISallesPCornejo-OlivasM, et al.LRRK2: genetic mechanisms vs genetic subtypes. Handb Clin Neurol2023; 193: 133–154.
84.
MasottiBTombesiGParisiadouL, et al.LRRK2 And the fragile synapse: a molecular prelude to Parkinson's disease?Biochem J2025; 482: 20251028.
85.
DouDAikenJHolzbaurELF. RAB3 Phosphorylation by pathogenic LRRK2 impairs trafficking of synaptic vesicle precursors. J Cell Biol2024; 223: 20240321.
86.
ChenCHeQTombesiG, et al.Leucine-rich repeat kinase 2 impairs the release sites of Parkinson's disease vulnerable dopamine axons. bioRxiv2025: 1–51. doi:https://doi.org/10.1101/2025.08.28.672006
87.
SternSLauSManoleA, et al.Reduced synaptic activity and dysregulated extracellular matrix pathways in midbrain neurons from Parkinson's disease patients. NPJ Parkinsons Dis2022; 8: 103. 20220810.
88.
SkitevaOYaoNSitziaG, et al.LRRK2-G2019S Mice display alterations in glutamatergic synaptic transmission in midbrain dopamine neurons. J Neurochem2022; 161: 158–172. 20220227.
89.
SitziaGSkitevaOCherguiK. Neuronal firing and glutamatergic synapses in the substantia Nigra pars Reticulata of LRRK2-G2019S mice. Biomolecules2022; 12: 20221104.
90.
NguyenMKraincD. LRRK2 Phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson's disease. Proc Natl Acad Sci U S A2018; 115: 5576–5581. 20180507.
91.
HeatonGRLandeckNMamaisA, et al.Sequential screening nominates the Parkinson's disease associated kinase LRRK2 as a regulator of Clathrin-mediated endocytosis. Neurobiol Dis2020; 141: 104948. 20200517.
92.
Connor-RobsonNBoothHMartinJG, et al.An integrated transcriptomics and proteomics analysis reveals functional endocytic dysregulation caused by mutations in LRRK2. Neurobiol Dis2019; 127: 512–526. 20190405.
93.
GalperJDeanNJPickfordR, et al.Lipid pathway dysfunction is prevalent in patients with Parkinson's disease. Brain2022; 145: 3472–3487.
94.
SchechterMAtiasMAbd ElhadiS, et al.alpha-Synuclein facilitates endocytosis by elevating the steady-state levels of phosphatidylinositol 4,5-bisphosphate. J Biol Chem2020; 295: 18076–18090. 20201021.
95.
SongDYYuanLCuiN, et al.alpha-Synuclein induces deficiency in clathrin-mediated endocytosis through inhibiting synaptojanin1 expression. J Neurochem2023; 167: 461–484. 20231003.
96.
VargasKJColosiPLGirardiE, et al.alpha-Synuclein colocalizes with AP180 and affects the size of clathrin lattices. J Biol Chem2023; 299: 105091. 20230728.
97.
BanksSMLMedeirosATMcQuillanM, et al.Hsc70 Ameliorates the Vesicle Recycling Defects Caused by Excess alpha-Synuclein at Synapses. eNeuro2020; 7: 1–18. 20200131.
98.
CaoMWuYAshrafiG, et al.Parkinson sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron2017; 93: 882–896.e885.
99.
McPhersonPSGarciaEPSlepnevVI, et al.A presynaptic inositol-5-phosphatase. Nature1996; 379: 353–357.
100.
SongWZinsmaierKE. Endophilin and synaptojanin hook up to promote synaptic vesicle endocytosis. Neuron2003; 40: 665–667.
101.
SongPPengWSauveV, et al.Parkinson's disease-linked parkin mutation disrupts recycling of synaptic vesicles in human dopaminergic neurons. Neuron2023; 111: 3775–3788.e3777. 20230915.
102.
ChoudhryHAggarwalMPanPY. Mini-review: synaptojanin 1 and its implications in membrane trafficking. Neurosci Lett2021; 765: 136288. 20211009.
103.
Leno-DuranEArrabalLRoldanS, et al.Identification of SYNJ1 in a Complex case of juvenile parkinsonism using a multiomics approach. Int J Mol Sci2024; 25: 20240909.
104.
PengYJGengJWuY, et al.Minibrain kinase and calcineurin coordinate activity-dependent bulk endocytosis through synaptojanin. J Cell Biol2021; 220: 20211001.
105.
GaoXHuangZFengC, et al.Multimodal analysis of gene expression from postmortem brains and blood identifies synaptic vesicle trafficking genes to be associated with Parkinson's disease. Brief Bioinform2021; 22: 1–15.
106.
ChoongCJAguirreCKakudaK, et al.Phosphatidylinositol-3,4,5-trisphosphate interacts with alpha-synuclein and initiates its aggregation and formation of Parkinson's disease-related fibril polymorphism. Acta Neuropathol2023; 145: 573–595. 20230320.
107.
ZouLZhangXXiongM, et al.Asparagine endopeptidase cleaves synaptojanin 1 and triggers synaptic dysfunction in Parkinson's disease. Neurobiol Dis2021; 154: 105326. 20210304.
108.
PanPYSheehanPWangQ, et al.Synj1 haploinsufficiency causes dopamine neuron vulnerability and alpha-synuclein accumulation in mice. Hum Mol Genet2020; 29: 2300–2312.
109.
ChangDNallsMAHallgrimsdottirIB, et al.A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet2017; 49: 1511–1516. 20170911.
110.
TrempeJFChenCXGrenierK, et al.SH3 Domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell2009; 36: 1034–1047.
111.
CaoMMilosevicIGiovediS, et al.Upregulation of Parkin in endophilin mutant mice. J Neurosci2014; 34: 16544–16549.
112.
ArranzAMDelbroekLVan KolenK, et al.LRRK2 Functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci2015; 128: 541–552.
113.
WatanabeSMamerLERaychaudhuriS, et al.Synaptojanin and endophilin mediate neck formation during ultrafast endocytosis. Neuron2018; 98: 1184–1197.e1186.
114.
ImotoYXueJLuoL, et al.Dynamin 1xA interacts with endophilin A1 via its spliced long C-terminus for ultrafast endocytosis. EMBO J2024; 43: 3327–3357. 20240621.
115.
HuynhDPScolesDRNguyenD, et al.The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet2003; 12: 2587–2597. 20030812.
116.
WangYZhuYLiW, et al.Synaptotagmin-11 inhibits synaptic vesicle endocytosis via endophilin A1. J Neurosci2023; 43: 6230–6248. 20230720.
117.
LiWRWangYLLiC, et al.Synaptotagmin-11 inhibits spontaneous neurotransmission through vti1a. J Neurochem2021; 159: 729–741. 20211014.
118.
PihlstromLAxelssonGBjornaraKA, et al.Supportive evidence for 11 loci from genome-wide association studies in Parkinson's disease. Neurobiol Aging2013; 34: 1708 e1707–1713. 20121113.
119.
DongNXieZWeiA, et al.Compensatory synaptotagmin-11 expression conceals Parkinson's disease-like phenotypes in parkin knockout mice. Cell Commun Signal2025; 23: 61. 20250203.
120.
ShimuraHHattoriNKuboS, et al.Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet2000; 25: 302–305.
121.
YoshinoHLiYNishiokaK, et al.Genotype-phenotype correlation of Parkinson's disease with PRKN variants. Neurobiol Aging2022; 114: 117–128. 20220106.
122.
KitadaTAsakawaSHattoriN, et al.Mutations in the Parkin gene cause autosomal recessive juvenile parkinsonism. Nature1998; 392: 605–608.
123.
GladkovaCMaslenSLSkehelJM, et al.Mechanism of parkin activation by PINK1. Nature2018; 559: 410–414. 20180606.
124.
CreedRBRobertsRCFarmerCB, et al.Increased glutamate transmission onto dorsal striatum spiny projection neurons in Pink1 knockout rats. Neurobiol Dis2021; 150: 105246. 20201230.
125.
TripathiURoshIBen EzerR, et al.Upregulated ECM genes and increased synaptic activity in Parkinson's human DA neurons with PINK1/ PRKN mutations. NPJ Parkinsons Dis2024; 10: 103. 20240518.
126.
NgXYWuYLinY, et al.Mutations in Parkinsonism-linked endocytic proteins synaptojanin1 and auxilin have synergistic effects on dopaminergic axonal pathology. NPJ Parkinsons Dis2023; 9: 26. 20230215.
127.
RoosenDABlauwendraatCCooksonMR, et al.DNAJC Proteins and pathways to parkinsonism. FEBS J2019; 286: 3080–3094. 20190620.
128.
NgJCortes-SaladelafontEAbelaL, et al.DNAJC6 Mutations disrupt dopamine homeostasis in juvenile Parkinsonism-dystonia. Mov Disord2020; 35: 1357–1368. 20200530.
129.
AbelaLGianfrancescoLTagliattiE, et al.Neurodevelopmental and synaptic defects in DNAJC6 parkinsonism, amenable to gene therapy. Brain2024; 147: 2023–2037.
130.
VidyadharaDJSomayajiMWadeN, et al.Dopamine transporter and synaptic vesicle sorting defects underlie auxilin-associated Parkinson's disease. Cell Rep2023; 42: 112231. 20230314.
131.
DorienARoosenNLBonet-PonceL, et al.Mutations in Auxilin cause parkinsonism via impaired clathrin-mediated trafficking at the Golgi apparatus and synapse. bioRxiv2021: 1–35. doi:https://doi.org/10.1101/830802
132.
JacquemynJKuenenSSwertsJ, et al.Parkinsonism mutations in DNAJC6 cause lipid defects and neurodegeneration that are rescued by Synj1. NPJ Parkinsons Dis2023; 9: 19. 20230204.
133.
ChengXTangYVidyadharaDJ, et al.Impaired pre-synaptic plasticity and visual responses in auxilin-knockout mice. iScience2023; 26: 107842. 20230906.
134.
LeeDWZhaoXZhangF, et al.Depletion of GAK/auxilin 2 inhibits receptor-mediated endocytosis and recruitment of both clathrin and clathrin adaptors. J Cell Sci2005; 118: 4311–4321.
135.
LinAWGillKKCastanedaMS, et al.Chemical genetic identification of GAK substrates reveals its role in regulating Na(+)/K(+)-ATPase. Life Sci Alliance2018; 1: e201800118. 20181231.
136.
DumitriuAPachecoCDWilkJB, et al.Cyclin-G-associated kinase modifies alpha-synuclein expression levels and toxicity in Parkinson's disease: results from the GenePD study. Hum Mol Genet2011; 20: 1478–1487. 20110121.
137.
NagleMWLatourelleJCLabadorfA, et al.The 4p16.3 Parkinson disease risk locus is associated with GAK expression and genes involved with the synaptic vesicle membrane. PLoS One2016; 11: e0160925. 20160810.
138.
EgawaJArtaRKLemmonVP, et al.The cyclin G-associated kinase (GAK) inhibitor SGC-GAK-1 inhibits neurite outgrowth and synapse formation. Mol Brain2022; 15: 68. 20220726.
139.
ZhangDYuanCLiuM, et al.Deficiency of SCAMP5 leads to pediatric epilepsy and dysregulation of neurotransmitter release in the brain. Hum Genet2020; 139: 545–555. 20200204.
140.
LeeURyuSHChangS. SCAMP5 Mediates activity-dependent enhancement of NHE6 recruitment to synaptic vesicles during synaptic plasticity. Mol Brain2021; 14: 47. 20210304.
141.
ParkDLeeUChoE, et al.Impairment of release site clearance within the active zone by reduced SCAMP5 expression causes short-term depression of synaptic release. Cell Rep2018; 22: 3339–3350.
142.
CastermansDVoldersKCrepelA, et al.SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Hum Mol Genet2010; 19: 1368–1378. 20100112.
143.
JiaoXMorleoMNigroV, et al.Identification of an identical de Novo SCAMP5 missense variant in four unrelated patients with seizures and severe neurodevelopmental delay. Front Pharmacol2020; 11: 599191. 20201218.
144.
LiuHGeSLiuZ, et al. Deficiency of SCAMP5 causes Parkinson's disease due to loss of dopamine neurons. Hum Genet2025; 144: 1139–1158. 20251104.
145.
LeeUChoiCRyuSH, et al.SCAMP5 plays a critical role in axonal trafficking and synaptic localization of NHE6 to adjust quantal size at glutamatergic synapses. Proc Natl Acad Sci U S A2021; 118: 1–10.
146.
AsmerianHDiazAJXuH, et al.Synaptic vesicles that store monoamines and glutamate differ in protein composition. bioRxiv2025: 1–49. doi:https://doi.org/10.1101/2025.05.06.651945
147.
NakatsuFMessaMNandezR, et al.Sac2/INPP5F is an inositol 4-phosphatase that functions in the endocytic pathway. J Cell Biol2015; 209: 85–95.
148.
BlauwendraatCHeilbronKVallergaCL, et al.Parkinson's disease age at onset genome-wide association study: defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord2019; 34: 866–875. 20190407.
149.
CaoMParkDWuY, et al.Absence of Sac2/INPP5F enhances the phenotype of a Parkinson's disease mutation of synaptojanin 1. Proc Natl Acad Sci U S A2020; 117: 12428–12434. 20200518.
150.
IshikawaSTanakaYTakahashi-NikiK, et al.Stimulation of vesicular monoamine transporter 2 activity by DJ-1 in SH-SY5Y cells. Biochem Biophys Res Commun2012; 421: 813–818. 20120425.
151.
LeonieMHegerFGGubinelliF, et al.VMAT2 Dysfunction impairs vesicular dopamine uptake, driving its oxidation and α-synuclein pathology in DJ-1-linked Parkinson’s neurons. Sci Adv2026; 12: eadz5645.
152.
LukBMohammedMLiuF, et al.A physical interaction between the dopamine transporter and DJ-1 facilitates increased dopamine reuptake. PLoS One2015; 10: e0136641. 20150825.
153.
Canet-AvilesRMWilsonMAMillerDW, et al.The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A2004; 101: 9103–9108. 20040604.
154.
MeulenerMCXuKThomsonL, et al.Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A2006; 103: 12517–12522. 20060807.
155.
YueQLiXWuF, et al.Unveiling the role of DJ-1 protein in vesicular storage and release of catecholamine with nano/micro-tip electrodes. Angew Chem Int Ed Engl2020; 59: 11061–11065. 20200428.
156.
SkouLDJohansenSKOkarmusJ, et al.Pathogenesis of DJ-1/PARK7-mediated Parkinson's disease. Cells2024; 13: 20240206.
157.
KyungJWKimJMLeeW, et al.DJ-1 deficiency impairs synaptic vesicle endocytosis and reavailability at nerve terminals. Proc Natl Acad Sci U S A2018; 115: 1629–1634. 20180131.
158.
GoldbergMSPisaniAHaburcakM, et al.Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron2005; 45: 489–496.
159.
GiangrassoDMFurlongTMKeefeKA. Characterization of striatum-mediated behavior and neurochemistry in the DJ-1 knock-out rat model of Parkinson's disease. Neurobiol Dis2020; 134: 104673. 20191115.
160.
SmithLSchapiraAHV. GBA Variants and Parkinson disease: mechanisms and treatments. Cells2022; 11: 20220408.
161.
ShinerTMirelmanAGana WeiszM, et al.High frequency of GBA gene mutations in dementia with Lewy bodies among ashkenazi Jews. JAMA Neurol2016; 73: 1448–1453.
162.
LesageSBriceA. Role of Mendelian genes in “sporadic” Parkinson's disease. Parkinsonism Relat Disord2012; 18: S66–S70.
163.
VidyadharaDJBackstromDChakrabortyR, et al.Synaptic vesicle endocytosis deficits underlie cognitive dysfunction in mouse models of GBA-linked Parkinson's disease and dementia with Lewy bodies. Nat Commun2025; 16: 8484. 20250926.
164.
RoshITripathiUHusseinY, et al.Synaptic dysfunction and extracellular matrix dysregulation in dopaminergic neurons from sporadic and E326K-GBA1 Parkinson's disease patients. NPJ Parkinsons Dis2024; 10: 38. 20240219.
165.
Eva Rodríguez-TraverLMSCrespoCGonzález-BurgosI, et al.GBA1 mutations alter neuronal firing and structure, regulating VGLUT2 and CRYAB in dopamine neurons. bioRxiv2024: 1–59. doi:https://doi.org/10.1101/2024.08.05.606574
166.
Merino-GalanLZamarbideMBelloso-IguerateguiA, et al.Resilience of striatal synaptic plasticity over early structural adaptations in premotor parkinsonism. NPJ Parkinsons Dis2025; 11: 146. 20250603.
167.
ChiuWHKovachevaLMusgroveRE, et al.alpha-Synuclein-induced Kv4 channelopathy in mouse vagal motoneurons drives nonmotor parkinsonian symptoms. Sci Adv2021; 7: 20210310.
168.
MoceriSBauerleNHabermeyerJ, et al.Young human alpha synuclein transgenic (BAC-SNCA) mice display sex- and gene-dose-dependent phenotypic disturbances. Behav Brain Res2024; 460: 114781. 20231202.
169.
MatschkeLAKomadowskiMAStohrA, et al.Enhanced firing of locus coeruleus neurons and SK channel dysfunction are conserved in distinct models of prodromal Parkinson's disease. Sci Rep2022; 12: 3180. 20220224.
170.
Jone RazquinLDLH-GVaquero-RodríguezAGonzález-AseguinolazaG, et al.Locus Coeruleus α-Synuclein Overexpression Induces Prodromal Parkinsonian Features in Mice. bioRxiv2025: 1–48. doi:https://doi.org/10.1101/2025.11.15.688542
171.
ButkovichLMHouserMCChalermpalanupapT, et al.Transgenic mice expressing human alpha-synuclein in noradrenergic neurons develop locus ceruleus pathology and nonmotor features of Parkinson's disease. J Neurosci2020; 40: 7559–7576. 20200831.
172.
PhanLMillerDGopinathA, et al.Parkinson's paradox: alpha-synuclein's selective strike on SNc dopamine neurons over VTA. NPJ Parkinsons Dis2025; 11: 207. 20250711.
173.
ZhouZYePLiXH, et al.Synaptic potentiation of anterior cingulate cortex contributes to chronic pain of Parkinson's disease. Mol Brain2021; 14: 161. 20211106.
174.
TangCXChenJShaoKQ, et al.Blunt dopamine transmission due to decreased GDNF in the PFC evokes cognitive impairment in Parkinson's disease. Neural Regen Res2023; 18: 1107–1117.
175.
IemoloADe RisiMGiordanoN, et al.Synaptic mechanisms underlying onset and progression of memory deficits caused by hippocampal and midbrain synucleinopathy. NPJ Parkinsons Dis2023; 9: 92. 20230616.
176.
ChenLNagarajaCDanielsS, et al.Synaptic location is a determinant of the detrimental effects of alpha-synuclein pathology to glutamatergic transmission in the basolateral amygdala. Elife2022; 11: 20220701.
177.
GcwensaNZRussellDLLongKY, et al.Excitatory synaptic structural abnormalities produced by templated aggregation of alpha-syn in the basolateral amygdala. Neurobiol Dis2024; 199: 106595. 20240706.
178.
LiuXYWangKDengXH, et al.Amelioration of olfactory dysfunction in a mouse model of Parkinson's disease via enhancing GABAergic signaling. Cell Biosci2023; 13: 101. 20230603.
179.
RobertsBMDoigNMBrimblecombeKR, et al.GABA Uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat Commun2020; 11: 4958. 20201002.
180.
WilsonDM3rdCooksonMR Van Den, et al.Hallmarks of neurodegenerative diseases. Cell2023; 186: 693–714.
181.
DejanovicBShengMHansonJE. Targeting synapse function and loss for treatment of neurodegenerative diseases. Nat Rev Drug Discov2024; 23: 23–42. 20231127.
182.
BakkerAKraussGLAlbertMS, et al.Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron2012; 74: 467–474.
183.
BakkerAAlbertMSKraussG, et al.Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. Neuroimage Clin2015; 7: 688–698. 20150221.
184.
VosselKRanasingheKGBeagleAJ, et al.Effect of levetiracetam on cognition in patients with Alzheimer disease with and without epileptiform activity: a randomized clinical trial. JAMA Neurol2021; 78: 1345–1354.
185.
ZwierzynskaEPietrzakB. The impact of Brivaracetam on cognitive processes and anxiety in various experimental models. Pharmacol Rep2024; 76: 86–97. 20240105.
186.
Rubio-NazabalEMajoieMSchulzAL, et al.Health-Related quality of life and cognitive performance during 12-month adjunctive Brivaracetam treatment in patients with focal-onset seizures: a prospective, observational study in Europe. Neurol Ther2025; 14: 609–625. 20250220.