
Abstract
Parkinson's disease (PD) is marked by motor symptoms and often accompanied by mild cognitive impairment (PD-MCI), affecting up to 50% of patients and preceding PD dementia (PDD). Genetic factors may influence this progression, yet the underlying mechanisms remain unclear. This study investigated genetic factors influencing the progression from PD-MCI to PDD using polygenic risk scores (PRS). A genome-wide association study (GWAS) was conducted using data from the LANDSCAPE study. Multivariable Cox regression, Kaplan-Meier survival analysis, and concordance statistics assessed the relationship between PRS and PDD progression. No significant association was found between PD PRS and the risk of developing PDD.
Plain language summary
This study aimed to understand whether specific genetic factors can help predict if people with mild cognitive impairment (MCI) in Parkinson's disease (PD) will go on to develop dementia, a condition known as Parkinson's disease dementia (PDD). Parkinson's disease is a progressive disorder that affects movement and can also lead to memory and thinking problems in many people. While some research has shown that certain genes are linked to Parkinson's disease itself, it's unclear if these same genetic factors affect the likelihood of developing dementia. To explore this, we used polygenic risk scores (PRS), which combine information from various genes to estimate a person's genetic risk for a disease. We analyzed data from people with Parkinson's disease and mild cognitive impairment, examining whether their genetic profiles could predict dementia progression over time. Statistical models were used to compare the genetic risk scores with actual dementia outcomes. Our findings showed no strong link between the genetic scores and the progression to dementia, suggesting that current genetic markers may not effectively predict this outcome in Parkinson's disease. These results highlight the need for more complex approaches that consider additional factors beyond genetics, including lifestyle or environmental influences. This research underscores that the development of dementia in Parkinson's disease may involve many factors and that genetic risk scores, as they are currently understood, may not be enough to predict who will develop dementia.
Keywords
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by motor symptoms such as bradykinesia, rigidity, and (resting-)tremor.1,2 In addition, mild cognitive impairment in PD (PD-MCI) is a common and early manifestation, with up to 50% of PD patients experiencing some degree of cognitive decline.2–4 This condition often precedes dementia, which significantly impacts quality of life and the healthcare burden. Despite extensive research, the factors influencing the progression from PD-MCI to PD dementia (PDD) remain unclear.
Genetic factors have been implicated in the etiology and progression of many neurodegenerative diseases, including PD.5–10 Genome-wide association studies (GWAS) have identified numerous single nucleotide polymorphisms (SNPs) associated with PD susceptibility, providing insights into the genetic underpinnings of the disease.11–13 SNPs are the most common type of genetic variation and may serve as biomarkers for disease risk, prognosis, and therapeutic response.
While numerous studies have successfully identified genetic variants associated with PD onset and motor symptoms, the genetic basis of cognitive impairment within PD remains poorly understood. Previous research has highlighted several genetic loci linked to cognitive decline in the general population and Alzheimer's disease (AD), suggesting a possible overlap with PD.14,15 However, the distinct pathophysiological mechanisms underlying PD necessitate a focused investigation.
PD-MCI serves as a critical stage for potential intervention to prevent or delay the onset of PDD. Identifying biomarkers and genetic factors associated with this progression is crucial for developing targeted therapies and personalized medicine approaches. Understanding the genetic basis of cognitive impairment in PD through GWAS could reveal critical biological pathways and potential targets for therapeutic intervention.
In this context, we conducted a comprehensive GWAS to investigate how genetic profiles influence the disease progression from PD-MCI to PDD. Therefore, we calculated the polygenic risk scores (PRS), which are derived from an individual's genotype profile and appropriate GWAS data and are used to estimate an individual's genetic susceptibility to an attribute or disease. or trait. To evaluate a potential association between PRS and PDD progression, we applied multivariable Cox regression models, Kaplan–Meier (KM) survival analysis, and concordance statistics were used to evaluate a putative link between the PRS and the progression to PDD and to determine whether SNPs influence the transition from PD-MCI to PDD.
Methods
Preparation of the LANDSCAPE SNP dataset
In this study, the data from the LANDSCAPE study for PD, which included 385 individuals within 7 years of follow-up, were used. 16 The genetic data was obtained with the GSA shared CUSTOM_24+ v1.0 array (Illumina). 364 samples passed the quality control, which was performed as previously described. 17 Briefly, samples with a genotyping rate below 98%, excess heterozygosity, and sex discrepancies were excluded. Additionally, we excluded SNPs with a call rate below 98% and significantly deviated from Hardy‒Weinberg equilibrium, indicated by a p value < 1 × 10−6. Principal component analysis (PCA) was performed via PLINK 1.9 (https://www.cog-genomics.org/plink/) for population stratification analysis. Finally, genetic coverage was maximized by imputation were imputed with the TOPMED panel (https://imputation.biodatacatalyst.nhlbi.nih.gov/) on the hg38 genome build. SNPs with an imputation quality ≤ 0.3 were excluded from the dataset. Rare variants with a minor allele frequency (MAF) < 0.02 were excluded from the analyses.
Calculation of the Alzheimer's disease PRS in the LANDSCAPE cohort
To calculate the AD PRS, information from two datasets, base and target data, was applied. The base data included the 83 independent risk SNPs extracted from the largest GWAS on Alzheimer's disease by the European Alzheimer & Dementia Biobank (EADB). 18 The target data were the same 83 SNPs from the LANDSCAPE genetic data, obtained as described above. The cumulative genetic susceptibility for PD was evaluated by aggregating the effects of the 83 GWAS-derived susceptibility loci.
Calculation of the Parkinson's disease PRS in the LANDSCAPE cohort
To calculate the PD PRS, the 90 independent SNPs extracted from the largest GWAS of PD were used as base data. 19 The target data were the same 90 SNPs from the LANDSCAPE dataset.
Calculation of the Parkinson's disease PRS via genome-wide survival studies in the LANDSCAPE cohort
Recently, a longitudinal GWAS including 3821 people living with PD identified a novel synaptic locus and 9 independent SNPs associated with disease progression. 20 To calculate PDD PRS, we used those 9 SNPs as base data and PD patient data from the LANDSCAPE study as target data for calculating the PRS.
Analysis
The association of each PRS with progression to dementia was performed by Cox-regression models and visualized using KM survival analysis. Only samples from the LANDSCAPE cohort with PD and PD-MCI (N = 322) at baseline were included in the analysis. Two models were applied: Model 1 tested the PRSs effects on time to conversion to dementia, and Model 2 tested PRSs effects on time to conversion to dementia adjusted by sex, age, Mini-Mental State Examination (MMSE), disease duration, and 3 PCAs. PRSs were stratified in tertiles (high, medium, and low), and their effects on time to conversion to PDD were visualized by KM survival curves. These analyses were also performed including only LANDSCAPE samples with PD-MCI (N = 153). Similar results were observed (data not shown).
Effect size calculation (Cohen's d)
To quantify the magnitude of group differences in polygenic risk scores (PRS), Cohen's d was computed as a measure of effect size. The pooled standard deviation was used to standardize the mean differences. Effect sizes were classified as small (0.2), medium (0.5), or large (0.8).
Post hoc power analysis
A post hoc power analysis was conducted to determine the achieved statistical power for detecting differences in PRS values between groups. The analysis was performed using the TTestIndPower function from the statsmodels package in Python, with a significance level of α = 0.05.
Sample size estimation
To estimate the required sample size for achieving 80% power, a prospective power analysis was conducted using the solve_power function from the statsmodels package in Python.
Results
We applied the PRS, which included European and top GWAS-independent SNPs, for survival analysis of PDD patients, adjusting for nongenetic risk factors. The demographic data of the investigated cohorts are shown in Table 1.
Demographic data of LANDSCAPE study for Parkinson's disease.
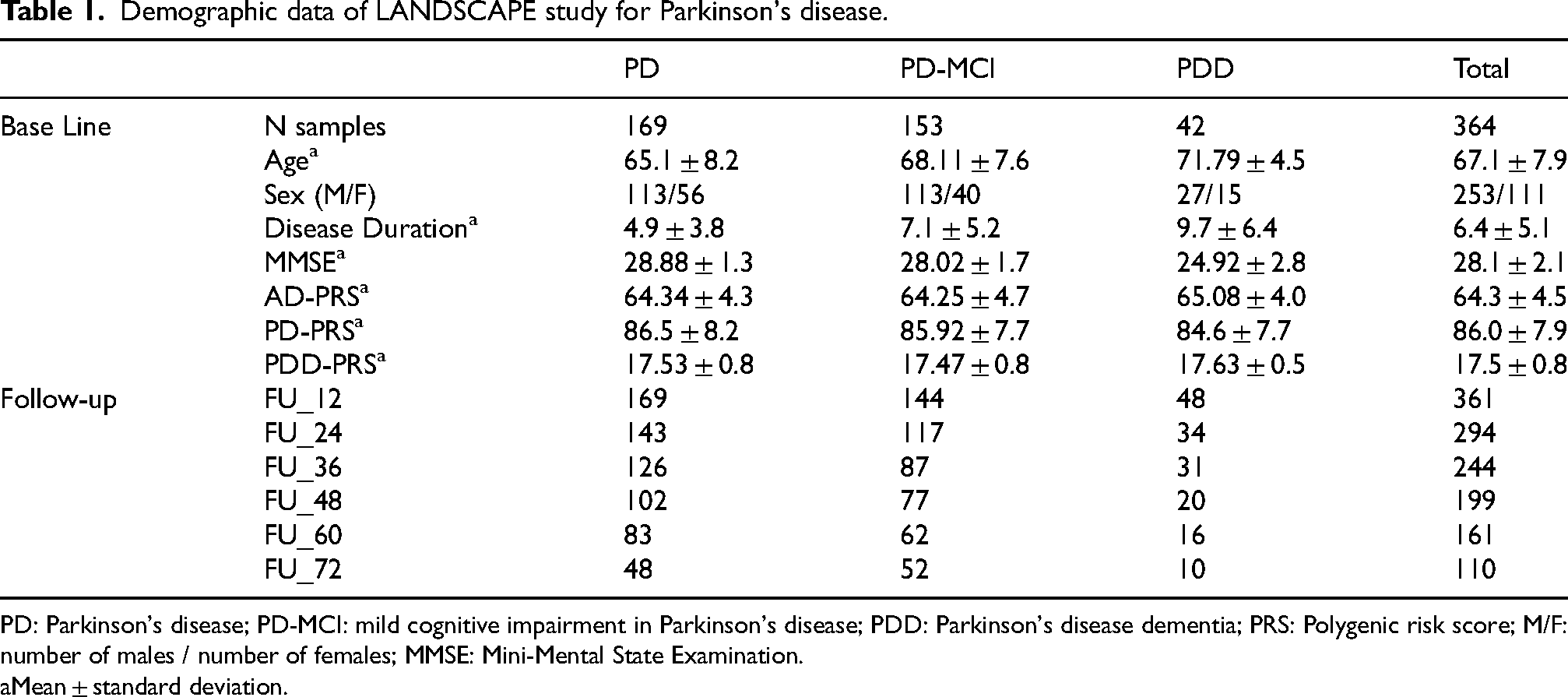
PD: Parkinson's disease; PD-MCI: mild cognitive impairment in Parkinson's disease; PDD: Parkinson's disease dementia; PRS: Polygenic risk score; M/F: number of males / number of females; MMSE: Mini-Mental State Examination.
Mean ± standard deviation.
Progression to PDD is independent of the AD PRS
No statistically significant association between the PRS and disease progression was found in either model, with a hazard ratio of 0.97 (95% CI: 0.93–1.07), as illustrated in Figure 1(a). The concordance statistics, indicating the goodness of fit for the model, revealed a score of 0.482 (SE = 0.047) for the PRS, which did not differ from chance. In the second model, age, MMSE score, and disease duration were significantly associated with disease risk. This evidence demonstrated that PRSs (hazard ratio = 0.99, 95% CI: 0.92–1.07) and related genetic variants are not significantly linked to disease progression. In the KM survival analysis, we plotted the time until individuals developed PDD against the PDD-free probability, categorized by PRS tertiles (Figure 1(a)). The median represents a 50% probability of developing PDD at an older age. This analysis revealed no significant difference in time to PDD development (p-value = 0.972) (Table 2).
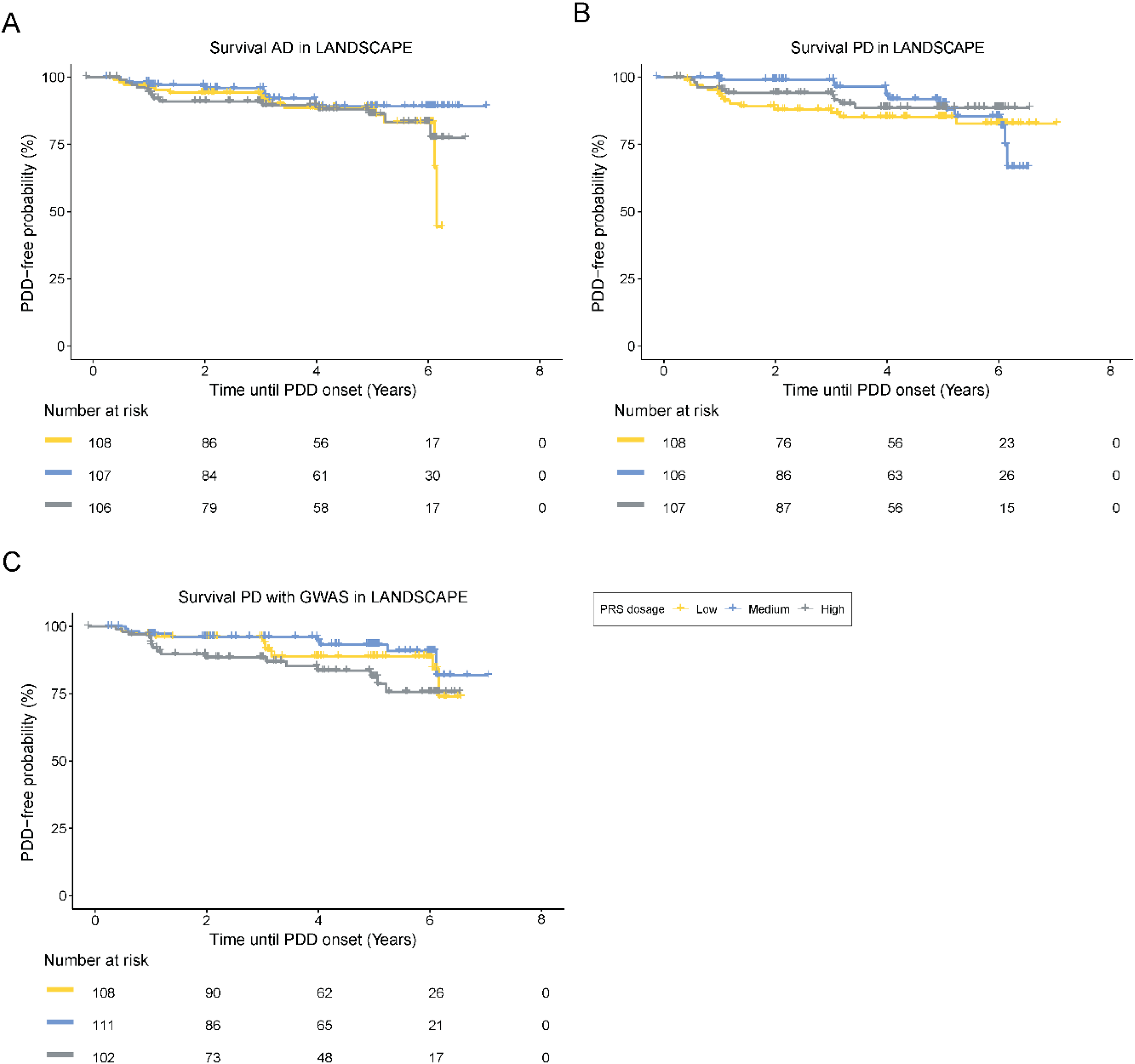
Kaplan–Meier survival curve for survival-free disease. (a) PRS of Alzheimer's disease. (b) PRS of Parkinson's disease. (c) PRS from PD with GWAS. AD: Alzheimer's disease, PD: Parkinson's disease, PDD: Parkinson's disease dementia, PRS: polygenic risk score.
Survival analysis of different PRS in the LANDSCAPE cohort.
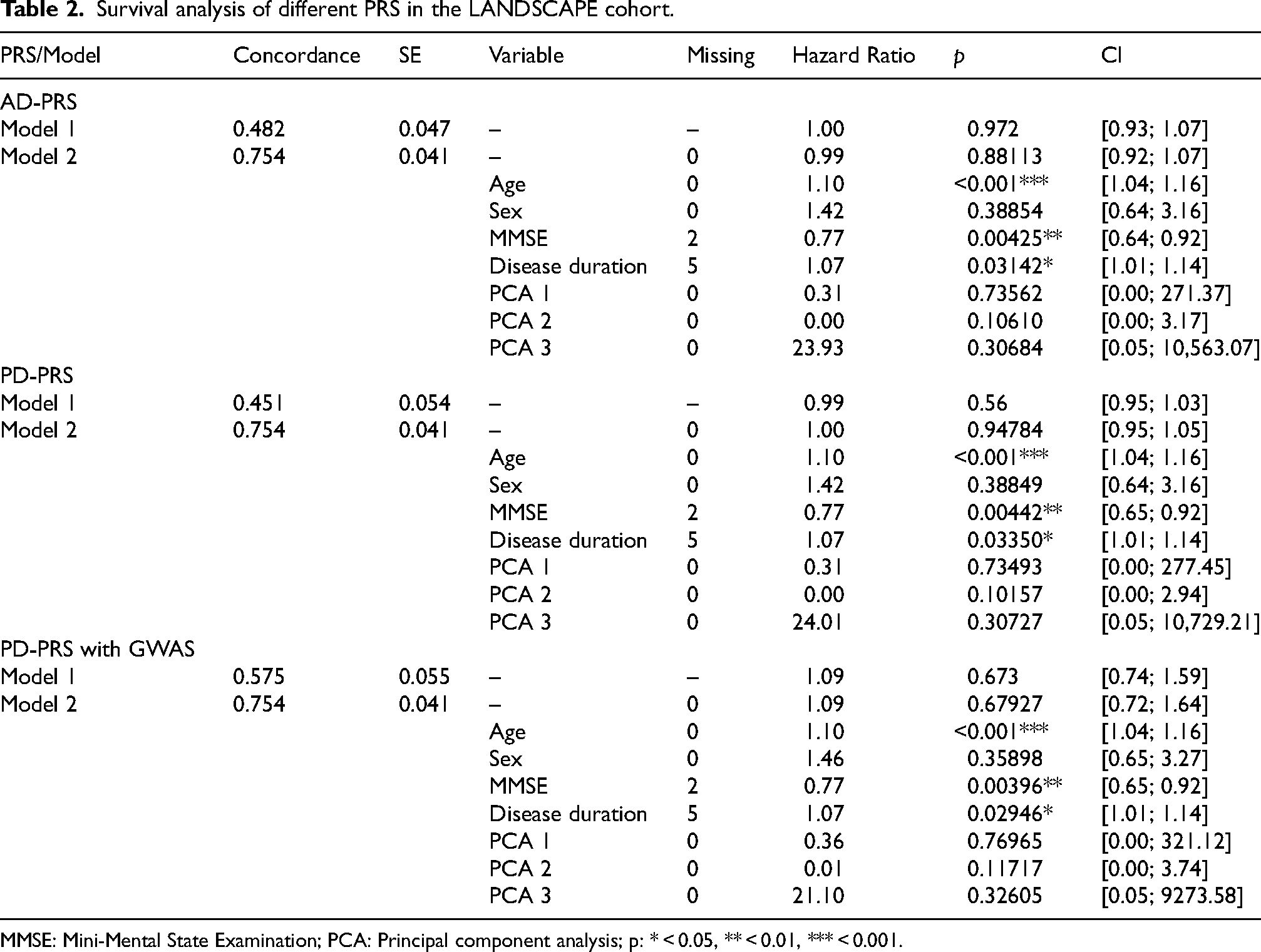
MMSE: Mini-Mental State Examination; PCA: Principal component analysis; p: * < 0.05, ** < 0.01, *** < 0.001.
Progression to PDD is independent of the PD PRS
We found no significant association between the PRS and disease progression in either model, with a hazard ratio of 0.99 (95% CI: 0.95–1.03). The concordance statistic for the PRS was 0.541 (SE = 0.054), indicating no significant predictive value. Cox regression analysis further confirmed that the PRS was not significantly associated with disease progression. For the KM survival analysis, we plotted the time until the onset of PDD against the PDD-free probability, categorized by PRS tertiles. As shown in Figure 1(b), the PRS did not significantly influence PDD development (p = 0.56) (Table 2).
Progression to PDD is independent of the GWAS-based the PD PRS
In this part of the analysis, we used nine independent SNPs from the LANDSCAPE study to calculate the PRS. The concordance statistic for the PRS was 0.575 (SE = 0.055), indicating that the PRS is not a better predictor for assessing disease progression (p = 0.673). For the KM survival analysis, we plotted the tertiles using the PRS from the PD GWAS, as shown in Figure 1(c). The analysis revealed no statistically significant association between the PRS and disease progression in either model, with a hazard ratio of 1.09 (95%-CI: 0.74–1.59). Sex had no effects on the associations (Table 2).
Statistical analysis of effect size, power, and sample size estimation
To evaluate the genetic influence on the progression PD-MCI to PDD, we calculated Cohen's d as a measure of effect size. The analysis was performed for the three PRS. The results indicated small effect sizes across all comparisons, with Cohen's d values of −0.171 for PD PRS, 0.178 for AD PRS, and 0.202 for GWAS PRS. These values suggest only minor differences in PRS distributions between groups.
Given the small effect sizes observed, a post hoc power analysis was conducted to assess the statistical power of the study. The power estimates for detecting differences between PD-MCI and PDD were 16.42% for PD PRS, 17.50% for AD PRS, and 21.04% for GWAS PRS, indicating that the study was underpowered for detecting small genetic effects.
To determine the necessary sample size for adequate statistical power, a prospective power analysis was performed. The analysis estimated that, to achieve 80% power at a significance level of α = 0.05, a minimum of 1250 participants per group for PD PRS, 1145 for AD PRS, and 898 for GWAS PRS would be required. These results highlight the need for larger cohorts or meta-analytic approaches to robustly detect small genetic effects associated with dementia progression in PD (Table 3).
Effect size, power, and sample size estimation.
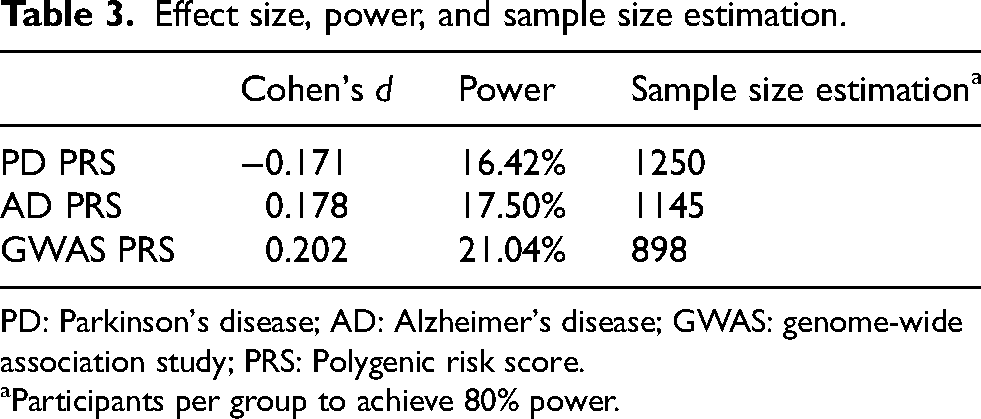
PD: Parkinson's disease; AD: Alzheimer's disease; GWAS: genome-wide association study; PRS: Polygenic risk score.
Participants per group to achieve 80% power.
Discussion
The results of our study suggest that neither the PRS derived from AD nor the PD-PRS summary statistics were associated with the development of PDD.
A possible explanation for our findings is the inherent complexity of the genetic architecture of PD. This complexity arises from numerous genetic and environmental factors influencing the disease, making it challenging for any single genetic estimator, such as the PRS, to capture the entire risk profile. While the PRS may estimate susceptibility to PD, it does not fully account for the diverse genetic contributions and multiple pathways involved in disease progression, including the development of dementia. The mechanisms driving PD onset may differ from those influencing its progression, complicating the ability of the PRS to serve as a reliable predictor across the disease spectrum. Therefore, more comprehensive models are needed to encompass the full range of genetic and non-genetic factors affecting PD to progression. Although our study did not identify a genetic correlation between PRS and the progression from PD-MCI to PDD, previous research has reported associations between PRS and other aspects of PD, such as REM sleep behavior disorder. 21 This highlights the limitations of current genetic predictors. On the one hand, these findings suggest a need for more nuanced approaches to understanding PDD progression, considering the complex interplay of genetic factors. On the other hand, they indicate that the transition to PDD may be influenced by modifiable environmental and lifestyle factors, reinforcing the importance of a holistic approach to PD risk and progression management. Our study has limitations, primarily due to limited statistical power resulting from a small sample size and the small effect sizes of genetic variants. This limitation reduced the sensitivity of our PRS approach. Nonetheless, while our results should be interpreted with caution, they align with previous research suggesting that genetic factors influencing AD and PD susceptibility do not necessarily contribute to dementia progression in PD patients. To further quantify the impact of these limitations, we conducted a post hoc power analysis, which revealed that the study was underpowered to detect small genetic effects. The observed effect sizes, as measured by Cohen's d, were small across all PRS comparisons, indicating only minor differences in PRS distributions between PD-MCI and PDD groups. Consequently, the achieved statistical power was substantially below the recommended threshold of 80%. A prospective power analysis further demonstrated that sample size of about 1000 and more participants per group would be required to achieve sufficient power. These findings emphasize the necessity of larger cohorts to reliably detect potential genetic effects on dementia progression in PD.
To improve the robustness of future findings, it is essential to include larger and more diverse cohorts, incorporate more comprehensive genetic data, and employ advanced statistical methodologies. Additionally, expanding the range of genetic variants analyzed and applying more sophisticated modeling techniques could help uncover subtle associations that are not captured by current PRS models. These approaches, combined with rigorous data processing and analysis pipelines, will be essential for minimizing biases and improving the reliability of conclusions drawn from genetic studies.
Conclusion
In conclusion, while our study revealed no significant association between AD or PD PRS and the risk of developing dementia, it emphasizes the complexity of genetic contributions to these neurodegenerative diseases. Our findings, consistent with previous research, highlight the importance of increasing statistical power and refining analytical methodologies to improve the detection of genetic risk factors. Future research should focus on larger and more diverse cohorts, comprehensive genetic datasets, and advanced analytical techniques to better understand and predict the genetic risk of developing neurodegenerative diseases.
Footnotes
Acknowledgements
We would like to express our gratitude to the patients and their relatives for their continuous support in obtaining the data in the LANDSCAPE study. D.B. has served as a consultant for UCB Pharma GmbH; Lilly Germany GmbH; Ottawa Hospital Research Institute; GT Gain Therapeutic SA, Switzerland. She received funding from Biohaven, BMBF, Deutsche Forschungsgemeinschaft (DFG), Else-Kröner-Forschungskolleg (EKFK), Hoffmann La Roche AG, Jan von Appen Stiftung, Lundbeck, Michael J. Fox Foundation (MJFF), UCB Pharma GmbH, EU and Novartis Pharma GmbH. G. D. has served as a consultant for Boston Scientific, Cavion and Insightec. He receives royalties from Thieme publishers. He receives funding from the German Research Council (SFB 1261, T1) and private foundations. A.S. has received funding from the Deutsche Forschungsgemeinschaft (German Research Association) and the Helmholtz-Association outside the present study. He has received honoraria for presentations/advisory boards/consultations from Esteve, Desitin, Lobsor Pharmaceuticals, STADA, Bial, RG Gesellschaft, Zambon, NovoNordisk and AbbVie outside the present study. He has received royalties from Kohlhammer Verlag and Elsevier Press. He serves as an editorial board member of Stem Cells International.K.W. receives funding from the Deutsche Forschungsgemeinschaft (German Research Association) and STADAPHARM GmbH outside the present study. He has received honoraria for presentations/advisory boards/consultations from BIAL, Indorsia, Boston Scientific and STADAPHARM GmbH, outside the present study. He has received royalties from Thieme Press and Elsevier Press. He serves as an editorial board member of Wileys “Parkinson's Disease”, “Behavioural Neurology” and PLOSone. R.D. has participated in industry-sponsored research projects from Lilly, Roche; served as a consultant for Lilly, Roche, Eisai, Novo Nordisk; received honoraria for scientific presentations from AbbVie, Bayer Vital, Lilly, Eisai, Schwabe, Roche; received publication royalties from Kohlhammer and Thieme; and is an inventor on patents from Philipps University Marburg for immunization in neurodegenerative diseases. The LANDSCAPE study was funded by the German Ministry for Education and Research (BMBF; project number 01GI1008C).
ORCID iDs
Ethical considerations
The study was conducted in compliance with the Helsinki Declaration. The study protocol was approved by the Ethics Committee of Philipps University Marburg (approval No. 178/07) and subsequently by the local ethics committees of the participating centers.
Consent to participate
All patients provided informed consent before enrollment in the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has received support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking (Aetionomy [grant number 115568]), and the Department of Neurology, University of Bonn.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The LANDSCAPE study was funded by the German Ministry for Education and Research (BMBF; project number 01GI1008C). The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data and genetic material can be provided on reasonable request by the corresponding author.