
Abstract
Background
While LRRK2 and GBA1 variants are associated with Parkinson's disease (PD), most carriers will not develop the disease.
Objective
To test if polygenic risk score (PRS) modifies disease risk and phenotypes in LRRK2 G2019S carriers, GBA1 carriers, and non-carriers (NC).
Methods
We genotyped 786 participants using Illumina's NeuroBooster-array (NBA) and sequenced the genome of 244, all of Ashkenazi ancestry (AJ), and calculated PRS to test its effects on clinically- and biologically-defined disease risk and phenotypes (n = 715). Among LRRK2 G2019S PD, we tested PRS association with α-synuclein seed-amplification-assay (n = 11). We used the PPMI and AMP-PD databases as validation cohorts.
Results
In clinically-defined PD, PRS significantly modified disease risk in GBA1 carriers and in NC (p = 0.033 and p < 0.0001, respectively), and demonstrated a trend in LRRK2 G2019S carriers (p = 0.054), with similar effect sizes (OR = 1.55, 1.62, and 1.49, respectively). PRS association with PD risk in LRRK2 was primarily driven by the rs7938782-A risk allele, replicated in AMP-PD (268 AJs LRRK2 G2019S carriers). PRS and age-at-onset were negatively correlated in NC (p < 0.0001). NBA GBA1 genotype calls failed at GBA1 L483P and c.115 + 1G > A mutations. False negative call rate of 10.2% was observed for the imputed GBA1 N409S carriers.
Conclusions
PRS contributes to PD risk across different genotypes. The genetic and epigenetic role of rs7938782 in LRRK2 PD risk should be further explored. Future PRS models should be tailored to specific genotypes to better understand penetrance and phenotypes. Furthermore, models predicting PD defined biologically rather than clinically may further identify genetic risk factors for synucleinopathies.
Plain language summary
Parkinson's disease (PD) is a genetically complex condition, caused by genetic and environmental risk factors. While specific mutations in the GBA1 and LRRK2 genes are major risk factors, most carriers will not develop PD. Here we examined why some carriers develop PD and others do not. Although environmental factors may influence this risk, we hypothesized that additional genetic risk factors play a role in disease development. Specifically, we tested if 86 known PD risk factors, analyzed as a polygenic risk score (PRS), increase PD risk in carriers, and affect disease phenotypes. This was tested in 715 unrelated individuals (PD patients and non-PD individuals), all of Ashkenazi Jewish ancestry. We split the cohort into 3 genetic groups: carriers of LRRK2 G2019S, carriers of risk variants in GBA1, and non-carriers (NC) of LRRK2 G2019S or GBA1 variants. Publicly available databases were used for validation. We found that elevated PRS (carrying many disease risk factors) was associated with increased PD risk in all three groups, but the effect was small. One risk factor (rs7938782-A) was significantly associated with PD risk in LRRK2 G2019S carriers but not in GBA1 carriers or NC, and its biological role should further be explored. Higher PRS was significantly associated with an earlier age-at-motor-symptoms-onset in NC. These findings indicates that PD risk across different genetic groups is affected by a combination of many changes in the genome. Future PRS models should be developed to better assess PD risk for different genetic backgrounds.
Introduction
Parkinson's disease (PD) is a complex heterogeneous neurodegenerative disease, with genetic risk factors identified across the genome. 1 Mutations in the LRRK2 and GBA1 genes are relatively common, specifically in Ashkenazi Jews (AJ) with PD, of whom up to 34% are carriers. 2 However, the majority of LRRK2 and GBA1 mutation carriers do not manifest PD throughout their lifetime.3–6 The model, which suggests that disease modification and level of penetrance are partially influenced by the contribution of single nucleotide variants (SNVs) residing within the PD-risk loci, was assessed and confirmed in several studies and in specific subgroups of genetic background (LRRK2 carriers, GBA1 carriers, and those who do not carry risk variants in GBA1 or LRRK2).4,7,8 The combined effect is expressed as a polygenic risk score (PRS) and can be calculated using different models. 9
Here we tested the association of PRS with PD, and its effect on different disease characteristics, including age-at-motor-symptoms-onset (AAO), hyposmia, and cognitive performance. We stratified the analyses to three genetic groups: LRRK2 G2019S carriers, GBA1 carriers, and non-carriers of the tested mutations. Moreover, given recent initiatives to biologically define and stage PD based on biological diagnosis of α-synuclein disease (NSD;10–12), we ran the analyses twice, defining PD clinically, and using a biological definition of PD. As cerebrospinal fluid (CSF) α-synuclein aggregation was previously reported in more than 93% of all GBA1-PD and idiopathic PD, but only in about 67% of LRRK2-PD, 13 we tested the association of PRS with CSF α-synuclein aggregation in LRRK2-PD patients. We further compared the performance of the Illumina NeuroBooster Array (NBA) genotyping accuracy of GBA1 and LRRK2 risk mutations to whole genome sequencing.
Methods
Terminology, abbreviations, and definitions of the different subgroups, used in this study are specified in Supplemental Table 1.
Cohorts
This study included 788 participants of the BEAT-PD study, a natural history study, which set out to characterize patients with PD, carriers of LRRK2 G2019S and GBA1 mutations, as well as non-carriers. In addition, the study included unaffected- first or second- degree- relatives of PD patients (non-manifest relatives, will be named NM-R hereafter), and unaffected controls with no family history of PD, as previously described.14,15 Briefly, patients were recruited if they were of AJ origin (self-reported), diagnosed as affected with PD based on the Movement Disorder Society (MDS) clinical diagnostic criteria for PD, 16 and were at Hoehn and Yahr stages 1–2. 17 All participants were examined at the Laboratory for Early Markers of Neurodegeneration (LEMON) at the Tel Aviv Sourasky Medical Center, Israel, and diagnosis was determined by movement disorders specialists. University of Pennsylvania Smell Identification Test (UPSIT) and Montreal Cognitive Assessment (MoCA) scores were collected as previously described.18,19 Percentiles of UPSIT scores were calculated and normalized to age and sex.
This study also included additional 244 participants (228 PD patients, 6 NM-R, and 10 unaffected controls who did not carry the LRRK2 G2019S mutation or any of the tested GBA1 mutations), who were examined at the Neurological Institute in Tel Aviv Sourasky Medical Center, and had their complete genome sequenced at 30X-read-depth coverage (Supplemental Figure 1).
All samples were genotyped for the LRRK2 G2019S mutation, and nine GBA1 pathogenic variants (p.Leu29AlafsTer18 c.115 + 1G > A, p.V433L, p.L483P, p.R535H, p.N409S, 370Rec (defined haplotype is described in 20 ), p.E365K, p.T408M), and SMPD1 L302P mutation, as previously described 2 (Supplemental Material). Individuals who carry the N409S, E365K, or T408M GBA1 variants and not the LRRK2 G2019S mutation are named GBA1(3) carriers hereafter. Individuals who did not carry any of these 10 mutations were categorized as non-carriers (NC).
As validation cohorts, we used the Parkinson's Progression Markers Initiative (PPMI) genetic LRRK2 and GBA1 arms (Tier 1 data: openly available from PPMI, downloaded between 08 November 2023 and 21 April 2024); To study the role of PRS in NSD-risk, we used data obtained from PPMI (Tier 3) on 29 October 2024 upon request, after approval by the PPMI Data Access Committee. For up-to-date data please visit the PPMI website at https://www.ppmi-info.org/). In addition, we used the Accelerating Medicines Partnership Parkinson's Disease (AMP-PD) data of AJ 21 to evaluate the rs7938782 variant (n = 268) (Supplemental Material).
High-throughput genotyping
Seven hundred and eighty-eight samples were genotyped on the Illumina NeuroBooster Array (NBA, beadchip 20042459, Illumina Global Diversity, https://www.medrxiv.org/content/10.1101/2023.11.06.23298176v2) using standard protocols. The array includes over 1.9 million markers from the Illumina Global Diversity Array and more than 95,000 Neurological disease-oriented and population specific variants, including several hundred known mutations in PD-related genes (https:\\github.com/GP2code/Neuro_Booster_Array).
Whole-genome-sequencing (WGS) was carried out for 244 AJ participants. Of them 221 were sequenced as previously described. 22 Briefly, sequencing was done with DNBSEQ technology at the BGI facility, China, with an average 30X-depth-coverage. Paired-end reads (each of 100 bp length) were aligned to the human reference genome (GRCh38 build) using the BWA tool, 23 and Genome Analysis Toolkit (GATK4) 24 was used for variant calling. An additional 23 PD patients were whole-genome-sequenced at 30X-depth-coverage on Illumina NovaSeq6000 platform (PCR-free libraries), with paired-end reads (150 bp length each) and secondary analysis was done using Illumina DRAGEN software with human reference genome GRCh38 build.
CSF collection and α-synuclein seed amplification assay (αs-SAA)
CSF was collected from a subset of participants of the BEAT-PD study (n = 99) by a neurologist through a lumbar puncture when the participants were either in the lateral recumbent position or sitting upright. CSF was extracted with a 21-gauge atraumatic needle, collected in 15-mL conical tubes, and centrifuged at 2000 Xg at 25° C for 10 min. The supernatant was collected into a conical tube, aliquoted into 0.5 mL, and frozen at −80° C. CSF aliquots were shipped with dry ice to Biogen, Inc and stored. αS-SAA was conducted and analyzed at Amprion as previously described. 13
Imputation of NBA-called variants and PRS calculation
The NBA genotype data of 788 samples were processed using a standard pipeline for imputation (Supplemental Material). PRSs with dosage were calculated in plink, using the imputed genotypes of the PD-risk loci reported by Nalls et al. 1 and averaged by the numbers of alleles (https://www.cog-genomics.org/plink/). As the analyses were done in a stratified manner, separately for each of the three subgroups, the PRS was calculated using 86 SNVs, omitting the LRRK2 and GBA1 variants: rs34637584, LRRK2 G2019S; rs76763715, GBA1 N409S; rs35749011, linked to GBA1 E365K; and rs114138760, linked to GBA1 T408M.
Estimated relatedness, principal component analyses, and statistical analyses
Estimated relatedness and principal component analyses (PCA) were performed with SNP & Variation Suite V.8.9.0 (Golden Helix, Inc) using either imputed genotypes or WGS genotypes (Supplemental Material). R (v4.3.2) was used to normalize PRS and PCA components (PC1 and PC2) as z scores (Z-PRS, Z-PC1, and Z-PC2, hereafter) and to run all statistical analyses. Stratified logistic and linear regression analyses are specified in Table 1. Differences in PRS between early onset PD (AAO ≤ 50) and late onset PD (AAO ≥ 70), and αS-SAA results of 11 LRRK2 G2019S PD patients in our cohort, were tested using the Mann-Whitney U-test. Number of individuals for each test are specified in Supplemental Tables 2 and 3, and additional statistics information is included in Supplemental Methods.
Statistical models used in analyses. TLVMC: Tel Aviv Sourasky Medical Center; PPMI: Parkinson's Progression Markers Initiative.
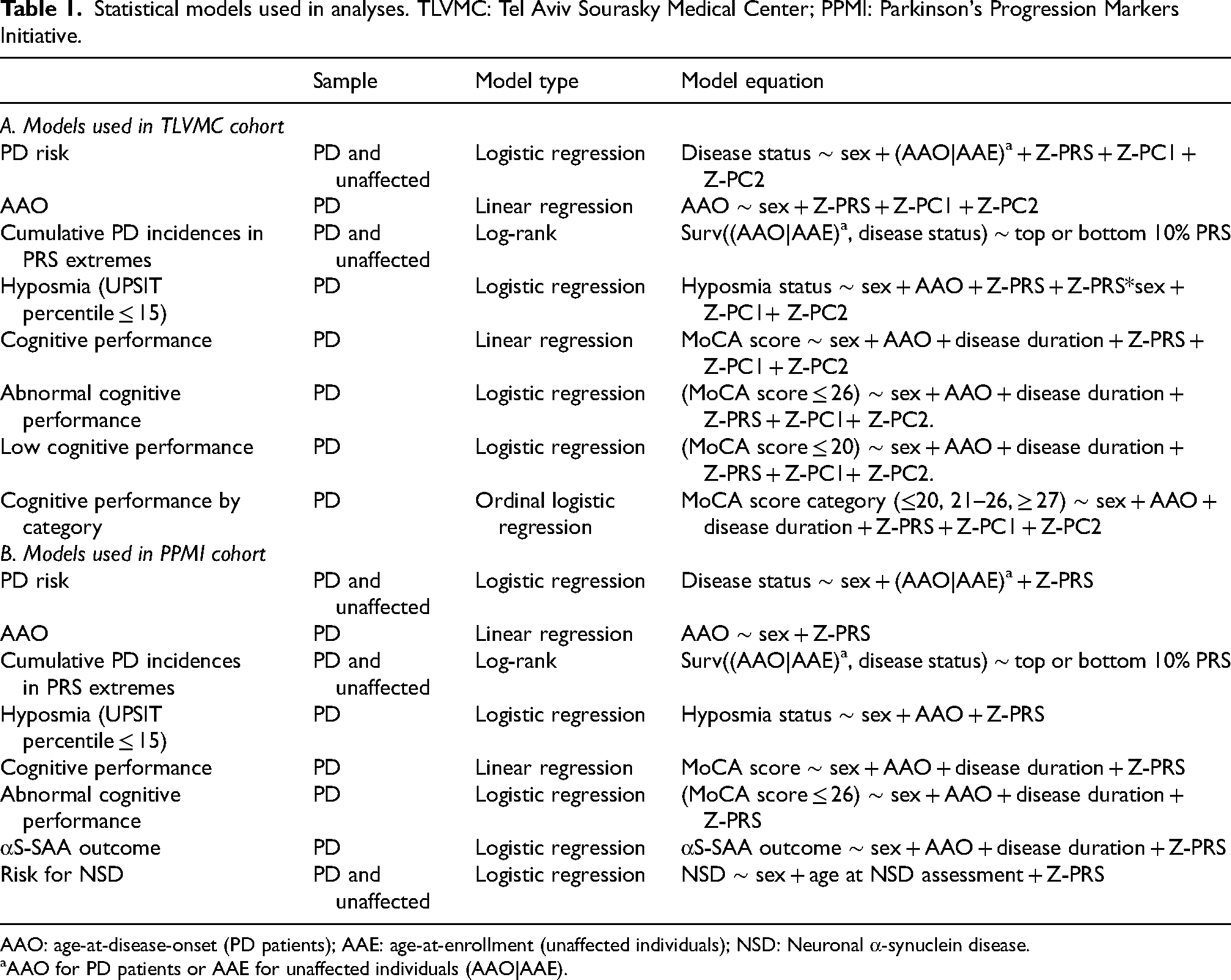
AAO: age-at-disease-onset (PD patients); AAE: age-at-enrollment (unaffected individuals); NSD: Neuronal α-synuclein disease.
AAO for PD patients or AAE for unaffected individuals (AAO|AAE).
Standard protocol approvals, registrations, and patients consents
All participants provided informed consent before entering the study. All DNA samples were coded and tested in an anonymous manner. The Institutional and Israeli National Supreme Helsinki Committees (IRBs) for Genetics Studies approved the study protocols and the informed consent.
Results
PD-risk and PRS
Plot of unadjusted PRS among PD patients and unaffected individuals showed that the mean PRS was higher in PD patients in all three groups: LRRK2 G2019S carriers, GBA1(3) carriers, and non-carriers (Figure 1A). Logistic regression analysis showed that among LRRK2 G2019S carriers, + 1 SD of PRS had a trend of association with PD-risk (OR = 1.49, 95% CI: 0.99–2.23, p = 0.054, Table 2). Among GBA1(3) carriers and NC, + 1 SD of PRS showed a significant association with PD-risk (OR = 1.55, 95% CI: 1.04–2.31, p = 0.033, and OR = 1.62, 95% CI: 1.28–2.05, p < 0.0001, respectively; Table 2). The latter association was significant also when comparing to NC NM-R, separately from NC Controls (OR = 1.66, 95% CI: 1.27–2.17, p = 0.0002, and OR = 1.67, 95% CI: 1.19–2.35, p = 0.0029, respectively, Table 2).
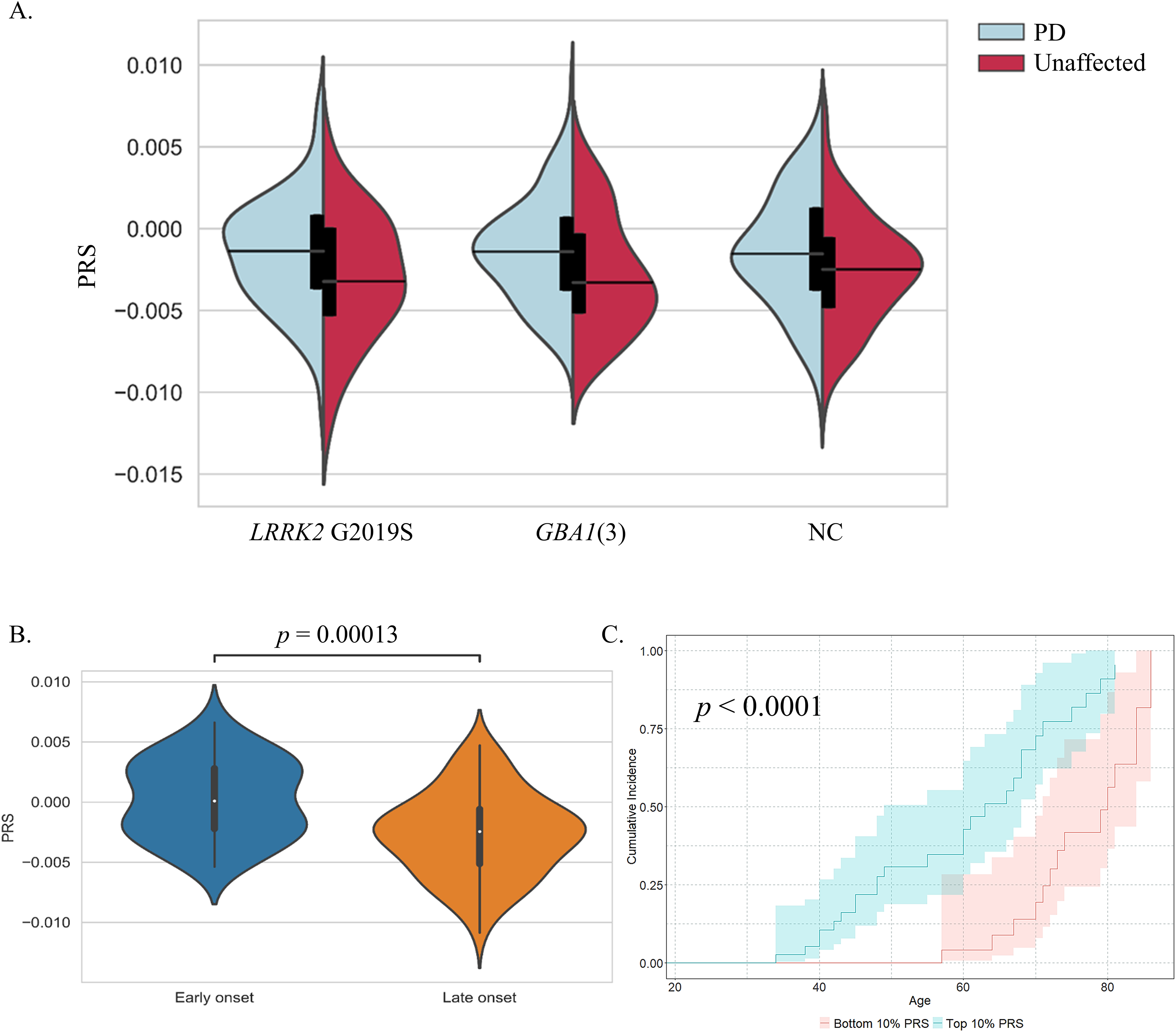
PRS effect on disease risk and age-at-onset. (A) Unadjusted PRS of PD and unaffected individuals in genetic groups. Mean PRS in PD patients was higher than in unaffected individuals among all three subgroups: LRRK2 G2019S carriers (nPD = 98, nunaffected = 46); GBA1 carriers of one of the three mutations N409S, E365K, T408M (GBA1(3), nPD = 149, nunaffected = 46); Non-carriers of the LRRK2 G2019S or any of the nine GBA1 mutations p.Leu29AlafsTer18, c.115 + 1G > A, p. V433L, p.L483P, p.R535H, p.N409S, 370Rec, p.E365K, p.T408M (nPD = 170, nunaffected = 206). (B) PRS in PD-NC with early AAO (AAO ≤ 50, n = 38) compared with late AAO (AAO ≥ 70, n = 72). (C) Cumulative PD incidence analysis in non-carriers among the top PRSs decile (blue) compared to the bottom PRSs decile (red). ntop 10% PRS = 38, of them 12 unaffected; nbottom 10% PRS = 38, of them 25 unaffected, p < 0.0001.
The effect of PRS on PD-risk in LRRK2 G2019S carriers, GBA1 carriers, and non-carriers.
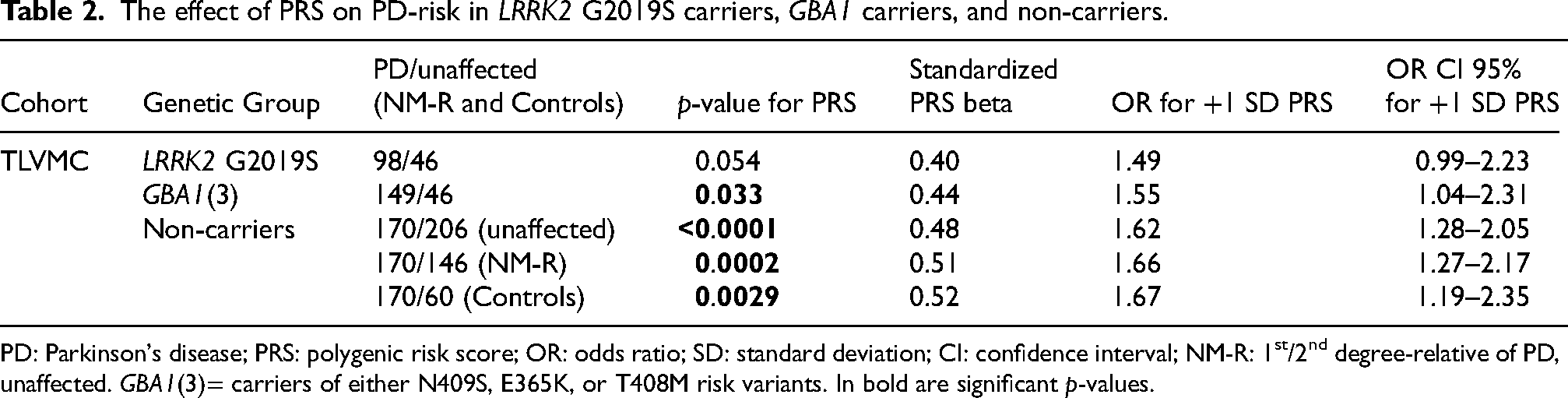
PD: Parkinson's disease; PRS: polygenic risk score; OR: odds ratio; SD: standard deviation; CI: confidence interval; NM-R: 1st/2nd degree-relative of PD, unaffected. GBA1(3)= carriers of either N409S, E365K, or T408M risk variants. In bold are significant p-values.
When testing each SNV's allele frequency in PD compared to non-PD, to identify a strong penetrance modifier, none of the SNVs was significantly different (after Bonferroni correction for multiple testing) in the GBA1(3) carriers and the non-carriers (Supplemental Table 4). However, in LRRK2 G2019S carriers, the rs7938782-A risk allele was significantly enriched in PD patients (Bonferroni p-value = 0.0100, Supplemental Table 4). This risk allele was also enriched in a secondary independent cohort of AJs carrying the LRRK2 G2019S mutation (AMP-PD, n = 268 unrelated individuals, of them 24 also carry GBA1(3) variant; OR = 1.64, 95% CI: 1.03–2.59, p = 0.0359). This allele, located in intron 1 of RNF141, is associated with a higher RNA level of AMPD3, with the highest effect size in the brain (GTEx, Supplemental Figure 2A). It is also associated with higher RNA level of MRVI1-AS1, again, with the highest effect size in brain (GTEx, Supplemental Figure 2B).
To assess the effect of PRS on disease defined by its biological αS-SAA results, we analyzed GBA1(3) carriers and non-carriers with conclusive αS-SAA results (Supplemental Table 3). The analyses of the low number of participants and the high overlap between clinical PD diagnosis and NSD status (determined by αS-SAA results) did not contribute additional insights (ORs > 1.0 for both GBA1(3) carriers and non-carriers, p > 0.05) beyond those offered by the PD model ran on a larger sample size.
Family history and PRS in unaffected NC individuals
Of the unaffected NC individuals, 60 were controls (with no family history of PD) and 146 had first-degree relative affected with PD (51 were related to LRRK2 G2019S-PD, 58 were related to GBA1-PD, 17 were related to NC-PD, and 20 were related to PD with an unknown genotype). The PRS of unaffected NC individuals with family history was not significantly different than those without family history (p = 0.1944). Moreover, among the unaffected NC individuals with family history, there were no significant differences in mean PRS between the three types of PD-genetic-family history (ANOVA, p = 0.52).
PRS and disease characteristics
Age-at-motor-symptoms-onset. In NC-PD patients, a significantly higher PRS was observed in early onset PD (AAO ≤ 50, n = 38) compared to late onset PD (AAO ≥ 70, n = 72) (Mann-Whitney U test, p = 0.00013, Figure 1B), but not among LRRK2 or GBA1(3) mutation carriers with PD (p = 0.0734 and p = 0.2804, respectively). Similarly, among NC individuals, the cumulative incidence of PD was significantly higher in those with a top 10% PRS compared to those with a bottom 10% PRS (p < 0.0001, Figure 1C). Linear regression analysis, adjusted for sex, PC1, and PC2, showed significance association of high PRS with lower AAO in NC (Beta = −4.875, p < 0.0001), but not in LRRK2 G2019S carriers or in GBA1(3) carriers (Beta = 0.745, p = 0.543 and Beta = −1.026, p = 0.224, respectively; Supplemental Table 5).
Hyposmia. We tested the effect of PRS on hyposmia (UPSIT percentile ≤ 15) in the three subgroups of PD patients. In LRRK2 G2019S PD, the PRS was significantly higher in females with hyposmia compared to females without hyposmia, but not in males (t-test, p = 0.0209 in females, p = 0.1744 in males, Supplemental Figure 3). To test the contribution of sex dependent PRS to hyposmia, while accounting for confounding parameters, we included an interaction term of PRS with sex in a logistic regression model (Table 1). Under this model, the probability of hyposmia was significantly higher in females compared to males for every +1SD of PRS change (OR (Z-PRS*sex)= 4.77, 95% CI: 1.20–18.89, p = 0.0262, Supplemental Table 6). When running the model on females and males separately, a trend for significance was observed in females (p = 0.0532) but not in males (p = 0.1715). No significance was observed in PD GBA1(3) or PD-NC (p = 0.5830 and p = 0.7779, respectively, Supplemental Table 6). We conclude that among LRRK2 G2019S PD patients but not among GBA1(3) or NC, elevated PRS significantly increases the risk of hyposmia in females compared to males.
Cognitive performance. When testing the effect of PRS on cognitive performance measured by MoCA scores, no significance was observed in any of the subgroups (p > 0.05), in all models (Supplemental Tables 7–9).
αS-SAA in LRRK2 G2091S PD carriers. In accordance with Siderowf et al. 13 all GBA1-PD (n = 16) and NC-PD (n = 14) were αS-SAA positive in CSF (100%) compared to 46% (6/13) in LRRK2-PD CSF, and therefore we assessed the effect of PRS on αS-SAA only in LRRK2-PD. Among unrelated LRRK2 G2019S PD, patients with positive αS-SAA in CSF (n = 6, all males, average AAO = 54.0) had a significantly higher PRS compared to those with a negative αS-SAA (n = 5, two females, average AAO = 66.4) (Mann-Whitney U-test, p = 0.0303, Supplemental Figure 4). All six αS-SAA positive patients (100%) had normalized UPSIT percentile score lower than 15th percentile (hyposmia), while only one out of the five αS-SAA negative patients (20%) was in the lowest 15th percentile (Fisher exact test, 2-tailed, p = 0.0152). This finding is in concordance with previous studies, associating α-synuclein aggregates with reduced UPSIT scores. 13
PRS association analyses in the PPMI cohort
Logistic regression analysis for disease risk showed similar significant results in NC as in our cohort (OR = 1.58, 95% CI: 1.31–1.92, p < 0.0001), but did not reach significance in LRRK2 G2019S carriers, or GBA1(3) carriers (OR = 1.02, CI: 0.74–1.42, p = 0.891, and OR = 1.29, CI: 0.87–1.92, p = 0.210, respectively). Concordant with the findings in our cohort, association of PRS with AAO was significant in NC, however with a smaller effect size than our cohort (Beta = −1.544, p = 0.0053, Supplemental Table 5). Albeit higher PRS in earlier AAO in both LRRK2 G2019S and GBA1(3) carriers, significance was not achieved (Beat = −1.622, p = 0.197, and Beat = −1.128, p = 0.430, respectively, Supplemental Table 5). When comparing PRS of early AAO to late AAO, no significance was observed in any of the groups, however, similarly to our cohort, the cumulative incidence of PD was significantly higher in those with a top 10% PRS compared to those with a bottom 10% PRS in NC (p = 0.0008, Supplemental Figure 5).
In the PPMI cohort, PRS did not significantly predict hyposmia or MoCA scores in any of the three groups (Supplemental Tables 6–9), nor did we observe an association with SAA status in LRRK2 G2019S PD patients (p = 0.3684).
Logistic regression was applied to assess if PRS predicts biologically defined PD in GBA1(3) (n = 117) and NC subgroups (n = 492) (Supplemental Table 3). We found that PRS was significantly associated with biologically defined PD among NC (OR = 1.299, 95% CI: 1.056–1.597, p = 0.0133), but not among GBA1(3) carriers (OR = 1.243, CI: 0.769–2.011, p = 0.375).
Neurobooster, imputation, and whole-genome-sequencing data overview and QC
The NBA genotypes of nine GBA1 mutations and the LRRK2 G2019S mutation, as well as their imputations, were compared to the well-established genotypes obtained previously by several different methods 2 (see Methods section and Supplemental Material). Results are summarized in Supplemental Table 10A. Call rate was 99.91%, with no false positive or false negative calls after excluding GBA1 L483P and GBA1 c.115 + 1G > A results (poor probes performance). Imputation results included only three of the GBA1 mutations (N409S, E365K, and T408M) and the LRRK2 G2019S mutation, with no false positive or false negative imputed calls for all except for N409S, which had 17 false- negative calls (10.2% error rate among the carriers, Supplemental Tables 10A and 11). Of the additional 86 SNVs building the PRS, imputation added missing genotypes to 48 SNVs (Supplemental Table 10B). Imputed genotypes for all 90 SNVs had a mean R2 of 0.996 (range 0.92–1.0, Supplemental Table 12), with 90% matched genotypes to WGS data (Supplemental Table 10C).
Of the 90 PD-risk SNVs, two are located within the GBA1 vicinity and are in linkage disequilibrium with GBA1 risk alleles: rs35749011 is linked to GBA1 E365K (located 73.8 Kb upstream), and rs114138760 is linked to GBA1 T408M (located 310.5 Kb upstream). Those SNVs are tagging the GBA1 risk variants, which are most likely the causal alleles for these GWAS signals. Interestingly, we found rare cases of individuals carrying recombinant alleles: one individual with a recombination event between GBA1 E365K and rs35749011 (carrier of the GBA1 mutation but not the linked SNV) and two individuals with recombination events between GBA1 T408M and rs114138760 (carriers of the linked variant but not the GBA1 mutation).
Discussion
We investigated the contribution of PRS to PD risk (clinically- or biologically- defined) among genetic groups, including LRRK2 and GBA1 carriers. As previously reported, we identified an association between PRS and PD status, supporting a polygenic nature of PD with additive effect of multiple risk loci, however the effect size was small. When we further explored differential effects of variants comprising the PRS, we identified the rs7938782-A risk allele in LRRK2 G2019S carriers, which was further confirmed using AMP-PD data, including 268 LRRK2 G2019S AJ carriers with and without PD.
The effect of rs7938782-A on AMPD3 and MRVI1-AS levels should further be explored in the context of PD. Increased expression of AMPD3 significantly reduces mitochondrial protein synthesis rate. 25 It is possible that lower rate of protein synthesis in the mitochondria in carriers of the rs7938782-A allele adds burden on mitochondrial function, an organelle known to be affected in LRRK2 G2019S PD patients. Interestingly, a recent study showed a significant lower RNA expression levels of MRVI1 (also known as IRAG1) in post mortem substantia nigra of PD patients compared to healthy controls. 26 MRVI1 encodes a protein which is a substrate of cGMP-dependent kinase-1 (PKG1) that can function as a regulator of IP3-induced calcium release. Defects in intracellular calcium homeostasis are known to play a role in PD. More specifically, it was shown that loss of PINK1 and Parkin leads to dysregulation of IP3-receptor activity, robustly increasing ER calcium release. 27 We can carefully hypothesize that higher levels of MRVI1-AS may indirectly affect the calcium release in the ER, however cellular studies should be carried out to understand the mechanism underlying this association to PD-risk.
As PD is associated with multiple risk factors, PRS can contribute to the prediction of PD and AAO in a genotype-dependent context. However, it should not be considered as a sole predictor. To increase PRS’ power of disease risk prediction, better models will need to be developed. These models should include gene-gene interactions, with different weights depending on genetic groups, as it was shown by us and others that some risk alleles have a higher effect size in a specific PD subgroup such as in the case of PARK16 locus risk alleles among LRRK2 G2019S carriers.4,8,28 Moreover, the model should be expanded to include epigenetics as well as environmental factors and family history.
Our results demonstrate that PRS calculated with PD-risk loci have limited predictive power for disease severity and phenotypes in GBA1 and LRRK2 G2019S carriers, including AAO, MoCA, and hyposmia. New models should be explored, as additional underlying processes may be contributing to these characteristics. Furthermore, models predicting PD defined biologically rather than clinically may further identify genetic risk factors for synucleinopathies, and for age of motor symptom development. Notably, the effect size of PRS in predicting AAO in the TLVMC cohort in NC individuals was approximately three times larger than that in the PPMI cohort. This difference may be attributed to the ethnic diversity present in the PPMI cohort, which is not present in our cohort.
Little is known about why some LRRK2 G2019S PD patients do not develop synucleinopathy. We hypothesized that higher PRS score would be associated with α-synuclein pathology as demonstrated by SAA positivity. While we observed such a signal in our cohort, we were not able to replicate it in the PPMI study. We suggest that larger cohorts are required to confirm or refute this observation. Additionally, other genomic models not restricted to PD-risk loci should be explored as they may refine the prediction model.
Given that the DNA of the study participants were analyzed both by NBA and other methods (WGS, Sanger Sequencing, TaqMan assays, GBA1 Array), we were able to evaluate the accuracy of NBA variant call in LRRK2 G2019S and GBA1. Due to the high homology between GBA1 and the adjacent pseudogene, GBAP1, specific variant calls, namely GBA1 L483P and GBA1-115 + 1G > A could not be obtained from NBA.
We identified rare cases of recombination events between the two GBA1 mutation variants (E365K and T408M) and the variants linked to them, represented in GWAS. Although these recombinant events are rare, it is important to be aware of them, and use the mutations themselves for PRS, if possible, and not the linked variants, when PRS is calculated using all 90 PD-risk variants.
The strengths of our study are the relatively large number of GBA1 and LRRK2 mutation carriers, genotyped with well-established and accurate methods, which allowed us to test PRS associations in a stratified manner, and to assess the new genotyping array, NBA. In addition, the cohort includes PD patients and first-degree-relatives of PD patients, all from the same AJ ancestry with detailed phenotypes. The major limitation of the study is the inclusion of only cross-sectional samples, and the relatively young age of the unaffected individuals that may still convert to PD, as well as the limited number of PD patients on whom a biological definition of PD was available (i.e., tested for αS-SAA).
In conclusion, we showed that PRS is significantly different between PD and unaffected individuals within each genetic subgroup, supporting the model that the LRRK2 and GBA1 mutations’ penetrance is modified by other genes within PD-risk loci, but not necessarily the same. Better models should be developed for each genetic subgroup, to create a more powerful marker for disease prediction. NBA is a cost effective genotyping platform, however its results should be used with caution, before and after imputation, and may require additional confirmations before clinical consultation or selection for GBA1 related trials.
Supplemental Material
sj-docx-1-pkn-10.1177_1877718X241310722 - Supplemental material for The effect of polygenic risk score on PD risk and phenotype in LRRK2 G2019S and GBA1 carriers
Supplemental material, sj-docx-1-pkn-10.1177_1877718X241310722 for The effect of polygenic risk score on PD risk and phenotype in LRRK2 G2019S and GBA1 carriers by Orly Goldstein, Shachar Shani, Mali Gana-Weisz, Nadav Elkoshi, Fergal Casey, Yu H Sun, Khyati Chandratre, Jesse M Cedarbaum, Cornelis Blauwendraat, Anat Bar-Shira, Avner Thaler, Tanya Gurevich, Anat Mirelman, Nir Giladi, Avi Orr-Urtreger and Roy N Alcalay in Journal of Parkinson's Disease
Footnotes
Acknowledgments
We would like to thank all of the participants who donated their time and biological samples to be a part of this study. This work was performed in partial fulfillment of the requirements for a PhD degree of Shachar Shani, Faculty of Medicine and Health Sciences, Tel Aviv University, Israel. Data used in the preparation of this article were obtained between 08 November 2023 and 21 April 2024, as well as on 29 October 2024, from the Parkinson's Progression Markers Initiative (PPMI) database (www.ppmi-info.org/access-data-specimens/download-data), RRID:SCR 006431. For up-to-date information on the study, visit www.ppmi-info.org. PPMI – a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, including 4D Pharma, Abbvie, AcureX, Allergan, Amathus Therapeutics, Aligning Science Across Parkinson's (ASAP) initiative, AskBio, Avid Radiopharmaceuticals, BIAL, BioArctic, Biogen, Biohaven, BioLegend, BlueRock Therapeutics, Bristol Myers Squibb, Calico Labs, Capsida Biotherapeutics, Celgene, Cerevel Therapeutics, Coave Therapeutics, DaCapo Brainscience, Denali, Edmond J. Safra Foundation, Eli Lilly, Gain Therapeutics, GE HealthCare, Genentech, GSK, Golub Capital, Handl Therapeutics, Insitro, Janssen Neuroscience, Jazz Pharmaceuticals, Lundbeck, Merck, Meso Scale Discovery, Mission Therapeutics, Neurocrine Biosciences, Neuropore, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi, Servier, Sun Pharma Advanced Research Company, Takeda, Teva, UCB, Vanqua Bio, Verily, Voyager Therapeutics, the Weston Family Foundation and Yumanity Therapeutics. Data used in the preparation of this article were obtained on Jan. 28, 2024 from the Accelerating Medicine Partnership® (AMP®) Parkinson's Disease (AMP-PD) Knowledge Platform. For up-to-date information on the study, visit ![]() . The AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson's (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson's Research; AbbVie Inc.; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences. ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of the U.S. Department of Health and Human Services.
. The AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson's (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson's Research; AbbVie Inc.; Pfizer Inc.; Sanofi US Services Inc.; and Verily Life Sciences. ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of the U.S. Department of Health and Human Services.
ORCID iDs
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Chaya Charitable Fund, Michael J Fox Foundation, and Biogen, Inc., This study was supported in part (CB) by the Intramural Research Program of the National Institutes of Health including: the Center for Alzheimer's and Related Dementias, within the Intramural Research Program of the National Institute on Aging and the National Institute of Neurological Disorders and Stroke.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The research study protocol and the informed consent form that were approved by the Institutional and National Supreme Helsinki Committees do not allow us to share patients’ information. However, if a specific request is made by a qualified investigator, anonymized data will be shared after approval by our IRB committee, and after an ethical agreement, MTA, is signed between the two institutions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.