
Abstract
Freezing of gait (FOG), a common, perplexing gait disorder observed in Parkinson's disease (PD), is a leading cause of injurious falls and contributes significantly to social isolation. Unlike other PD cardinal features, FOG appears to develop independently, and its heterogeneity presents challenges for both definition and measurement. The pathophysiological mechanisms underlying FOG remain poorly understood, limiting the development of effective treatments. Although the roles of specific, targetable biomarkers in FOG development remain unidentified, evidence suggests that it is likely multimodal, potentially involving extranigral transmitter circuits. The diversity of FOG phenotypes may also reflect underlying differences in pathophysiology. In this paper, we first present evidence that FOG may occur independently of dopaminergic influence. We then review an expanding body of research supporting the hypothesis that FOG arises from a dysfunctional pathophysiological feedback loop, involving norepinephrine (NE) depletion, neuroinflammation, and amyloid-β (Aβ) accumulation. This biological disruption occurs concurrently with, but distinct from, the primary dopaminergic pathology of PD. When they occur on the background of dopamine loss, the interactions between NE, Aβ, and inflammation, as observed in Alzheimer's disease models, may similarly play a critical role in the development of FOG in PD and could serve as pathobiological markers. The proposed changes in the pathophysiological loop might even precede its onset, highlighting the need for further investigation. A deeper understanding of the involvement of Aβ, NE, and inflammatory markers in FOG could pave the way for rapid clinical trials to test existing amyloid-clearing therapies and noradrenergic drugs in appropriate patient populations.
Plain language summary
Parkinson's disease (PD) is a progressive neurological disease associated with the loss of dopamine in the brain. Freezing of gait (FOG) is a complex abnormality of walking seen in people with PD where they feel like their feet are glued to the floor. It is common (affecting up to 60% of patients), a leading cause of injurious falls and contributes significantly to social isolation. It appears to develop and progress separately from other clinical symptoms of PD. Cognitive impairment is frequently associated with FOG. The cause is unknown, and treatment is limited. It is well known that there are several types of FOG suggesting that there might be more than one cause. There is also information suggesting that FOG, in some cases, may occur independent of the loss of dopamine that typically occurs in PD. In this paper we review information supporting that notion. Then, we develop our hypothesis of a different chemical pathway as a possible cause of FOG. This pathway involves the protein amyloid (too much in the brain) and the chemical norepinephrine (too little in the brain), both have been previously related to the development of Alzheimer's disease. We review the literature (including some of our work) that supports the possible role of this chemical pathway in the development of FOG. A deeper understanding of this chemical pathway in FOG could pave the way for the development of new effective drugs in appropriate patient populations.
Introduction
Freezing of gait (FOG) is an enigmatic gait disorder observed in Parkinson's disease (PD) and other forms of parkinsonism. It is defined as “Paroxysmal episodes where there is an inability to step effectively, despite attempting to do so.” (Jerusalem Workshop 2023). It is common in PD and poorly understood from the standpoint of pathophysiology. Treatment options are limited.1,2 FOG is a considerable public health burden worldwide, occurring in up to 26% of PD patients in the early stages of disease and then increasing over time to afflict >60% of patients at after 10 years of disease.3,4 This translates to approximately 4 million people with PD being afflicted. Similar figures are seen for atypical parkinsonism.5,6 It is a leading cause of injurious falls, and results in loss of independence with social isolation.1,7 A key feature of FOG is its clinical heterogeneity, which makes it a challenge to define, measure and treat. It has several clinical phenotypes; presenting with or without a complete cessation in the progression of the feet, cessation can be expressed with or without movement in the legs (most frequently known to be tremulous vs. akinetic; these movements, however, are variable in nature, not always tremulous). 8 It also is heterogeneous in triggers and response to levodopa. In between episodes, gait is also affected in that it often deteriorates in the moments before a FOG event. 9 FOG is also heterogeneous regarding associated non-motor features including executive dysfunction, psychosis and anxiety.10–12 The episodic nature and variability of response to dopaminergic medication of FOG has led to the conclusion that it is largely independent from other cardinal features of PD and likely has separate, currently unknown, pathophysiology. 13 The implication is that existing PD biomarkers, treatment targets, and even conceptual models are of little use for FOG.
The specific roles of targetable biomarkers leading to the development of FOG remain unknown because it appears to result from dysfunction in multiple systems or nodes beyond the dopaminergic deficit of PD. FOG heterogeneity goes beyond clinical features as there are wide-ranging findings related to physiological 14 and imaging research 15 which likely underly the varied motor and nonmotor phenotypes, and levodopa responsiveness. Such data further suggests that FOG in PD is likely multimodal including extranigral transmitter circuits. The diversity of FOG phenotypes may also reflect underlying differences in pathophysiology.
Discussion
Evidence that FOG can be non-dopaminergic
The first indicator for a possible role of extranigral circuits is evidence that FOG may be free from dopaminergic influence. Two lines of data support this notion. The first includes the occurrence of FOG in parkinsonian disorders without evidence of dopaminergic deficit. Fasano et al. 16 examined DaT SPECT using 123I-FP-CIT SPECT, and out of 13 cases with primary progressive FOG, five were normal. Those with normal and abnormal imaging had varied underlying etiologies. Factor et al. 17 performed 123I-β-CIT SPECT in 9 subjects with primary progressive FOG, and 7 were normal. These results indicate that dopaminergic deficit is not a necessary component for the development of FOG and pointing to a role for other extranigral systems. Further, one pathological report of a patient with primary progressive FOG found preserved substantia nigra. 18
The second line of evidence involves the existence of levodopa non-responsive FOG in PD. Several patterns of response have been reported by several investigators: (1) FOG which appears only in the “OFF” medication state, and disappears in the “ON” medication state (levodopa responsive FOG or OFF-FOG); (2) FOG that is present in both the “OFF” and “ON” states (levodopa unresponsive FOG or ONOFF-FOG); and (3) FOG present during “ON” state and absent in the “OFF” state (drug-induced FOG or ON-FOG or diphasic FOG).19–22 One study reported that 62% of FOG patients (self-reported) had OFF-FOG, 36% ONOFF-FOG, and 2% ON-FOG. 23 Another study found 40%, 57%, and 3%, respectively, in rigorously tested patients with levodopa challenges. 19
A rigorous assessment of these patterns was through a medication challenge study reported by McKay et al., 19 which conclusively demonstrated that FOG can persist even in the presence of adequate levodopa dosing. To elicit FOG in a clinical setting, subjects were assessed with the MDS-UPDRS-III as well as timed-up-and-go tests and 360° turns in a motion capture laboratory.24,25 Subjects were assessed in the practically defined “OFF” state, >12 h without levodopa, 26 and after supra-therapeutic levodopa doses (∼130% of the usual morning levodopa equivalent dose (LED)) to bring on a full “ON” state (>20% improvement in total MDS-UPDRS-III). 27 Serum levodopa levels were used to demonstrate adequate levodopa dosage and gut absorption. Despite clear levodopa responsiveness of their parkinsonism as a whole, sixteen subjects had levodopa unresponsive FOG (ONOFF-FOG) (Figure 1). Eighty percent of these had dyskinesias. Ten of these cases were levodopa unresponsive from the outset.
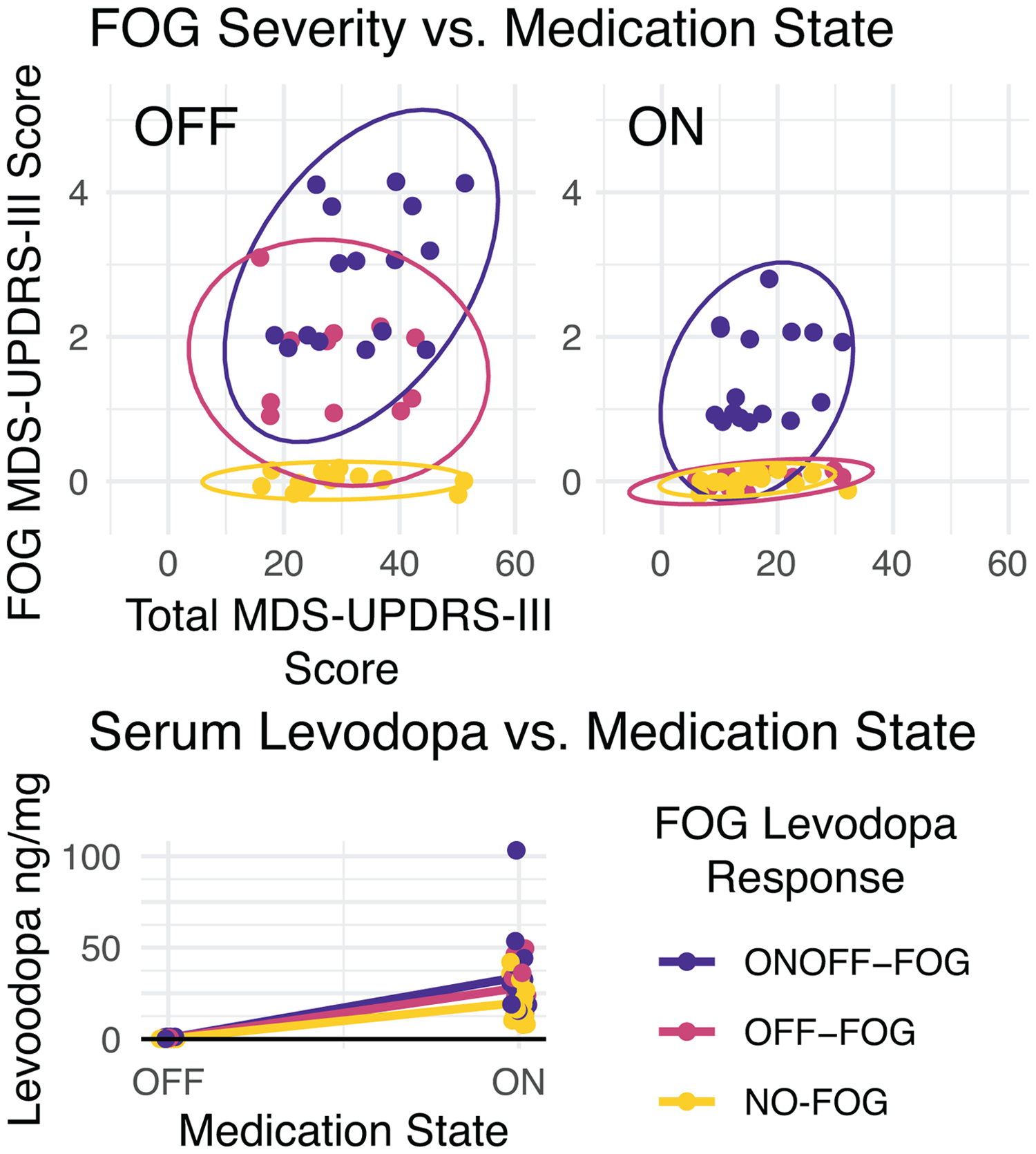
Clinical FOG scores and serum levodopa levels during a levodopa challenge paradigm. In the upper row, total motor symptom severity is shown on the x-axis and FOG score is shown on the y-axis. A small amount of jitter has been added for visualization. In individuals with “ONOFF” FOG (blue), FOG scores remain elevated even after levodopa doses causing significant improvements in overall motor score and significant increases in serum levodopa levels (lower row”) N = 42. Adapted from. 19
Hypothesis
It is possible that both levodopa unresponsive and levodopa responsive FOG develop on a background of a different, non-dopaminergic circuit dysfunction. There is a growing body of evidence supporting a role for norepinephrine (NE) depletion and associated markers as one such pathway. The NE circuit has been proposed to play a role in FOG through distinct compensatory networks involving cognitive, sensory and limbic systems. 28 It has been shown that loss of NE is associated with amyloid aggregation, at least in part, via inflammatory pathways, in Alzheimer's disease (AD). We suggest that these interactions, although with some variations, may also contribute to the development of FOG in PD patients on the background of dopamine depletion. Our central hypothesis is that FOG results from a dysfunction in a pathophysiological feedback loop incorporating NE depletion, neuroinflammation, and amyloid accumulation, and that occurs concurrently with, but distinct from, primary PD dopaminergic pathology.
NE has widespread distribution in the brain and is vital for arousal, attention, stress responses, mood, and cognition,29–31 all which relate to the development of FOG. The primary NE nucleus is the locus coeruleus (LC) which projects to subcortical and cortical regions, including frontal cortex, thalamus, hypothalamus, brainstem, amygdala, hippocampus, and cerebellum.32–36 A notable and complex link between NE, inflammation, and amyloid-β42 (Aβ42) has been characterized in animal models of AD (Figure 2), whereby NE protects against Aβ42 pathology. 37 Numerous studies have reported that noradrenergic lesions using the selective LC neurotoxin (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4)) exacerbates plaque deposition in mice overexpressing mutant human amyloid precursor protein (APP) and/or presenilin-1. 38 These effects of NE loss appear to have both inflammation-dependent and -independent components.
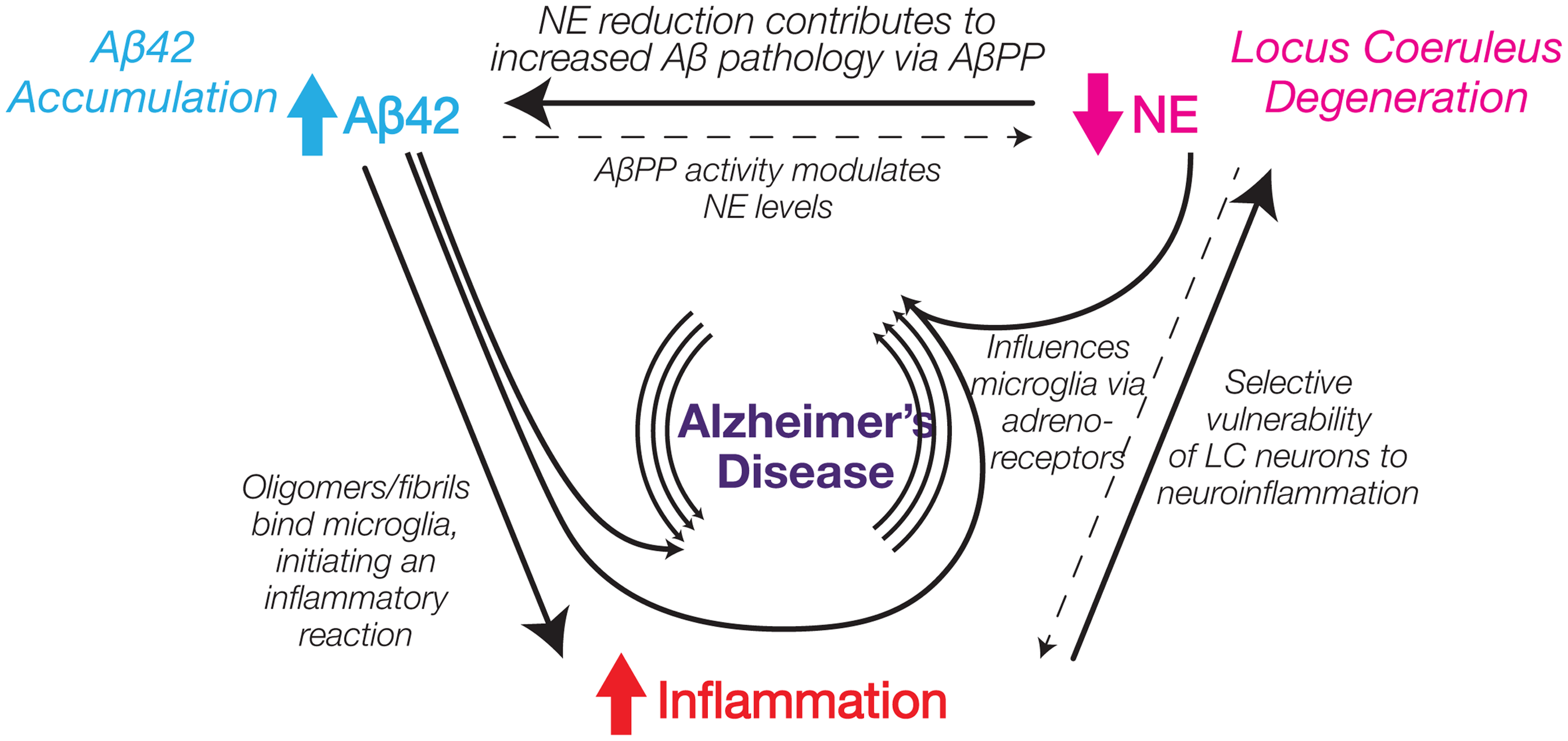
Schematic diagram illustrating interactions between multiple non-dopaminergic systems in Alzheimer's disease.
Pathological accumulation of Aβ42 is also a key driver of inflammation.39,40 Oligomers and fibrils bind to cell-surface receptors activating microglia and resulting in production of proinflammatory cytokines, chemokines, eicosanoids, and complement.39,40 This results in impaired clearance of Aβ42, loss of synapses, and neuronal death. Increased cytokine concentrations, by downregulation of expression of Aβ phagocytosis receptors, may be responsible for insufficient microglial phagocytic capacity leading to poor clearance of the amyloid. NE is a key modulator of neuroinflammation itself through influence of microglia function.41–43 Adrenergic receptors are expressed not only on neurons, but also microglia, and astrocytes. 42 The same DSP-4 lesions of the LC in AD mouse models that drive Aβ plaque burden also increase inflammation and neuronal damage.39,44 Among the most intriguing and convincing findings was that microglia, which typically engulf plaque material in APP transgenic mice, failed to do so following LC lesion, and this phenotype was rescued by restoring NE with the synthetic precursor L-3,4-dihydroxyphenylserine (L-DOPS; droxidopa), a drug used to treat FOG. 45
NE may also influence Aβ levels directly. In one study in non-human primates, injections of DSP4 accelerated the onset and incidence of AD-like pathology particularly in the prefrontal, temporal, and parietal cortices. 38 The reduction of NE contributed to increased Aβ pathology via altered APP processing.46,47 In addition, the amine oxidase activity of APP modulates systemic and local NE levels. 48 Adrenergic receptors have been shown to directly influence β- and γ-secretase activity, important in the metabolism of APP. 49 A recent cerebrospinal fluid (CSF) analysis demonstrated the complexity of the relationship between NE and Aβ. In 111 cognitively impaired patients, memory deficits were linked to the level of the NE principal metabolite 3-methoxy-4-hydroxyphenylglycol (MHPG) via a synergistic relationship with p-tau and Aβ42. 50 Hence, NE may be pivotal to understanding links between pathology and behavioral and cognitive deficits in AD.
NE and PD with FOG
There is evidence that NE and Aβ42 act as disease modifiers in PD explaining some phenotypic variation such as cognitive changes. The potential linkage of NE and Aβ42 to each other, as well as to cognitive decline and FOG in PD, suggests that they could serve as specific pathobiological markers for FOG. LC is often a site of significant early degeneration in PD and undergoes catastrophic degeneration later in the course of disease.51–54 It has been demonstrated that functional connections exist between LC degeneration and dopamine loss in the parkinsonian state.41,55 Studies in PD animal models and patients show that LC-NE loss may exacerbate PD motor symptoms, contribute to dopaminergic neuronal degeneration, and attenuate the therapeutic effects of levodopa,41,56–60 potentially involving neuroinflammation.60,61 NE loss may contribute to many PD clinical signs, including cognitive impairment and motor decline.55,62 In the 1980s, Droxidopa (formally known as L-DOPS), in a large clinical trial in Japan demonstrated a moderate to marked improvement in FOG in 25% of patients.63,64 However, these findings were controversial and have never been replicated despite two attempts to do so in smaller trials. 13 Nevertheless, the notion that degeneration of the LC, and the accompanying loss of NE in the brain, plays a role in FOG led to the approval of droxidopa to treat FOG in Japan. The NE transporter (NET) inhibitor atomoxetine, which increases extracellular levels of NE, has shown some promise in small studies for the treatment of FOG.65,66 Imaging studies have also supported the role of noradrenergic system in FOG. One examined aromatic 1-amino acid decarboxylase (AADC) activity in the LC using high-resolution positron emission tomography (PET) with an AADC tracer 6-[18F]fluoro-1-m-tyrosine (FMT, a marker for NE metabolism) in 40 PD patients (30 with FOG) and 11 age-matched healthy controls. The study demonstrated that the severity of FOG as measured by the FOG questionnaire correlated with the decrease of FMT uptake specifically in the LC irrespective of disease duration and the severity of other motor features (r = −0.69, p < 0.01), suggesting dysfunction of the noradrenergic circuit in FOG. 67 Another study examined NET binding via brain PET using the radioligand [11C]MeNER (2S,3S)(2-[α-(2-methoxyphenoxy)benzyl]morpholine) ([11C]MeNER) to evaluate changes in NET density associated with FOG. 28 Fifty-two patients were included and classified via rigorous levodopa challenge as PD-NoFOG (N = 16), PD-OFF-FOG (N = 10), PD-ONOFF-FOG (N = 21), and primary progressive (non-PD) FOG (N = 5). Levodopa-responsive FOG, specifically, was linked to reduced expression in multiple brain regions. The OFF-FOG group exhibited reduced whole brain NET binding compared to the No-FOG group (−16.8%, p = 0.021) (Figure 3). Region specific examinations demonstrated decreased NET expression in the left and right frontal and temporal lobes, thalamus, amygdalae, and LC, with the strongest effect observed in the right thalamus in the OFF-FOG group (p < 0.038). Linear regression analysis revealed a correlation between decreased NET binding in the right thalamus and more severe scores on the New FOG Questionnaire in the OFF-FOG group (p < 0.022). The noradrenergic regions of the thalamus represent limbic projections which have been previously linked to FOG28,68 and relate to known predictive risk factors such as depression and anxiety. 69 The reduced binding in OFF-FOG in particular is consistent the close relationship between NE and dopamine loss as previously noted. It is possible that in the OFF-FOG group there may be a dopaminergic pathway that is able to compensate for NET loss leading to FOG. Larger studies are required to investigate this hypothesis.
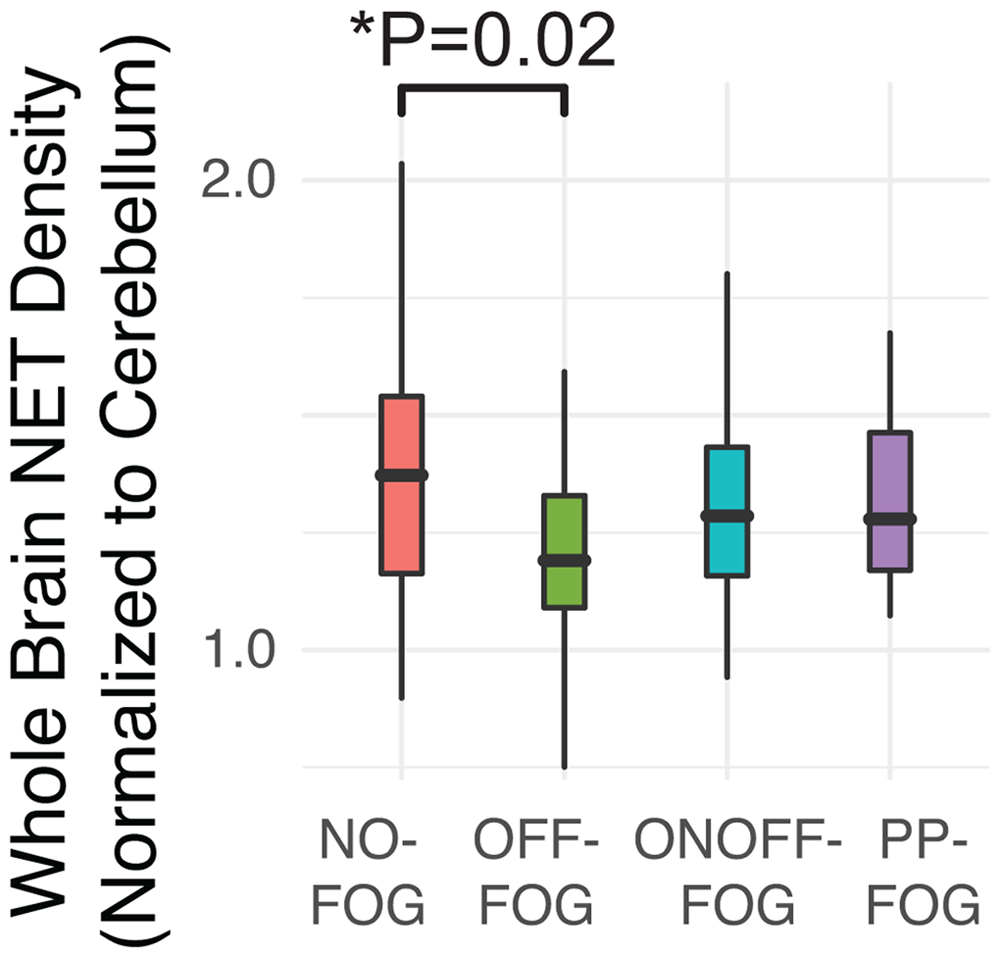
Variation in whole brain NET density across PD, FOG, and primary progressive FOG (PP-FOG). P-value reflects multivariate linear model controlled for PD duration and brain region. N = 60. Adapted from. 28
Amyloid, inflammatory markers, and PD with FOG
FOG is closely linked to cognitive decline and amyloid pathology, with studies showing lower CSF Aβ42 levels early in PD and elevated levels in advanced stages of those with FOG, suggesting a possible progression-related biomarker. FOG is generally found to be associated with cognitive decline, particularly executive dysfunction.10,11,70 Cognitive decline, in turn, in PD has been associated with abnormal (lower) CSF Aβ42. Imaging studies have demonstrated that cortical and subcortical amyloid aggregation is associated with poor performance in attentive-executive domains. This may identify amyloid-related cortical dysfunction in early PD. 71 In relation to development of FOG, an imaging study using Pittsburgh Compound B (PiB)-PET in 20 PD subjects with FOG performed multivariate logistic regression analysis and demonstrated that neocortical amyloid aggregation is an independent predictor of development of FOG. 72 A study of CSF markers (Aβ42, α-synuclein, total tau, phosphorylated tau181, and the calculated ratio of Aβ42 to total tau) at baseline in 393 newly diagnosed PD patients without FOG at baseline, from the Parkinson Progression Marker Initiative (PPMI), examined predictors of development of FOG. There was a median follow-up of 4.0 years. Cox proportional-hazards regression analyses demonstrated that only lower Aβ42 (∼8%) among the CSF biomarkers was associated with the ultimate development of FOG (hazard ratio 0.997, 95% confidence interval [CI] 0.996–0.999, p = 0.009). 73 In a more recent CSF study of a more advanced PD cohort, it was found that FOG is associated with elevated CSF Aβ42. The study included CSF samples collected from PD patients (12 with and 19 without FOG) and healthy controls (HC; N = 43). 74 Multivariate models adjusted for sex, disease duration, and multiple comparisons showed systematic changes in AD-related biomarkers. Specifically, Aβ42 was significantly elevated (p < 0.001) in PD-FOG (while in PD-NoFOG it was reduced) (Figure 4). To explore potential changes over time, additional multivariate regression models demonstrated a significant association (p = 0.02) between PD duration and CSF Aβ42 levels. Furthermore, after controlling for age and sex, a significant interaction (p < 0.01) between the PD-FOG and PD-NoFOG groups was observed. CSF Aβ42 was positively associated with PD duration in PD-FOG but negatively associated in PD-NoFOG, supporting the hypothesis of increasing Aβ42 over time in PD-FOG. This finding corroborates the PPMI data findings 73 as levels are low early on and rise over time.
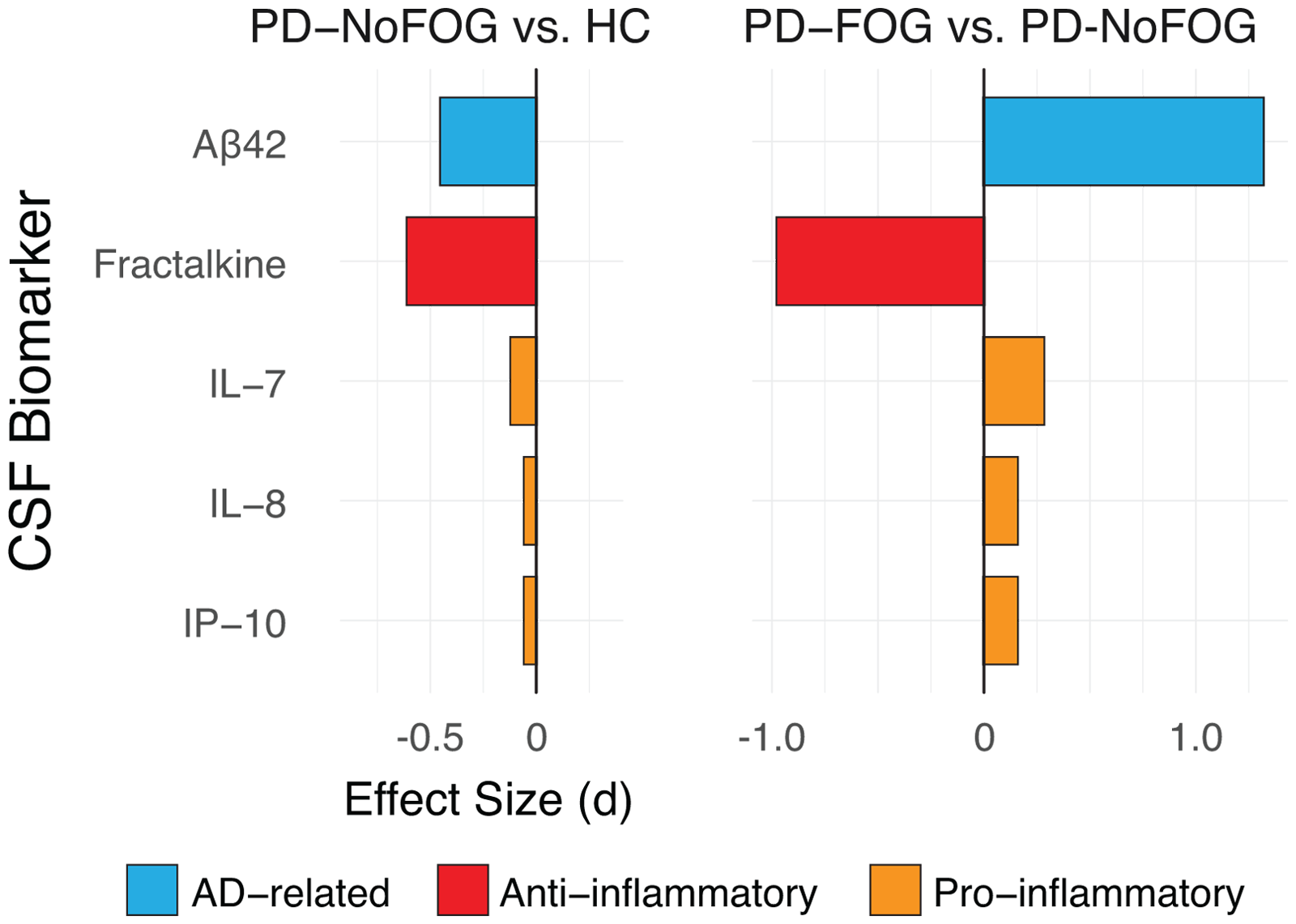
Contrasts in CSF Aβ, fractalkine, and inflammatory markers associated with PD and FOG. All effects sizes are shown as Cohens d. Modified from Hatcher-Martin, McKay et al., 2021.
Limited research on PD-FOG and inflammation reveals a dysregulated cytokine profile with reduced anti-inflammatory fractalkine in FOG cases, suggesting Aβ42 and fractalkine as potential indicators of FOG. The above discussed CSF study 74 also examined 10 inflammatory markers and found a dysregulated cytokine profile in PD patients with FOG compared to no FOG (Figure 4). The anti-inflammatory cytokine fractalkine was observed to be significantly reduced in PD FOG cases vs. NoFOG cases (p < 0.001). 74 In addition to the univariate analyses, a multivariate partial least squares discriminant analysis identified additional CSF inflammatory markers (IL-7, IL-8, IP-10) associated with FOG. Effect sizes for the contrasts in expression of these biomarkers across groups demonstrate decreased CSF anti-inflammatory markers, and increased CSF pro-inflammatory markers. Finally, a dual-tracer PET study with FEPPAVT for microglia and PIB for amyloid, demonstrated significant interactions between Aβ42 and activated microglia affecting cognitive performance on PD patients with MCI, more prominent in the earlier stage of disease. This further supports the connection of amyloid and inflammation in PD. 75
The PD CSF data discussed above demonstrates a trend seen in the AD models with regard to the relationship of CSF NE metabolite MHPG and the CSF Aβ42. As MHPG decreases, Aβ42 increases, linking these two nodes. Because MHPG is the principal metabolite of NE degradation in brain, one would predict that there would be an inverse relationship between MHPG and Aβ42 concentration in CSF from PD-FOG patients, and this is indeed the case. In a sample of N = 12 PD patients with FOG, these two quantities were inversely related (Pearson's r −0.32), providing support for the association between NE and Aβ42 in our overall hypothesis (Figure 5).
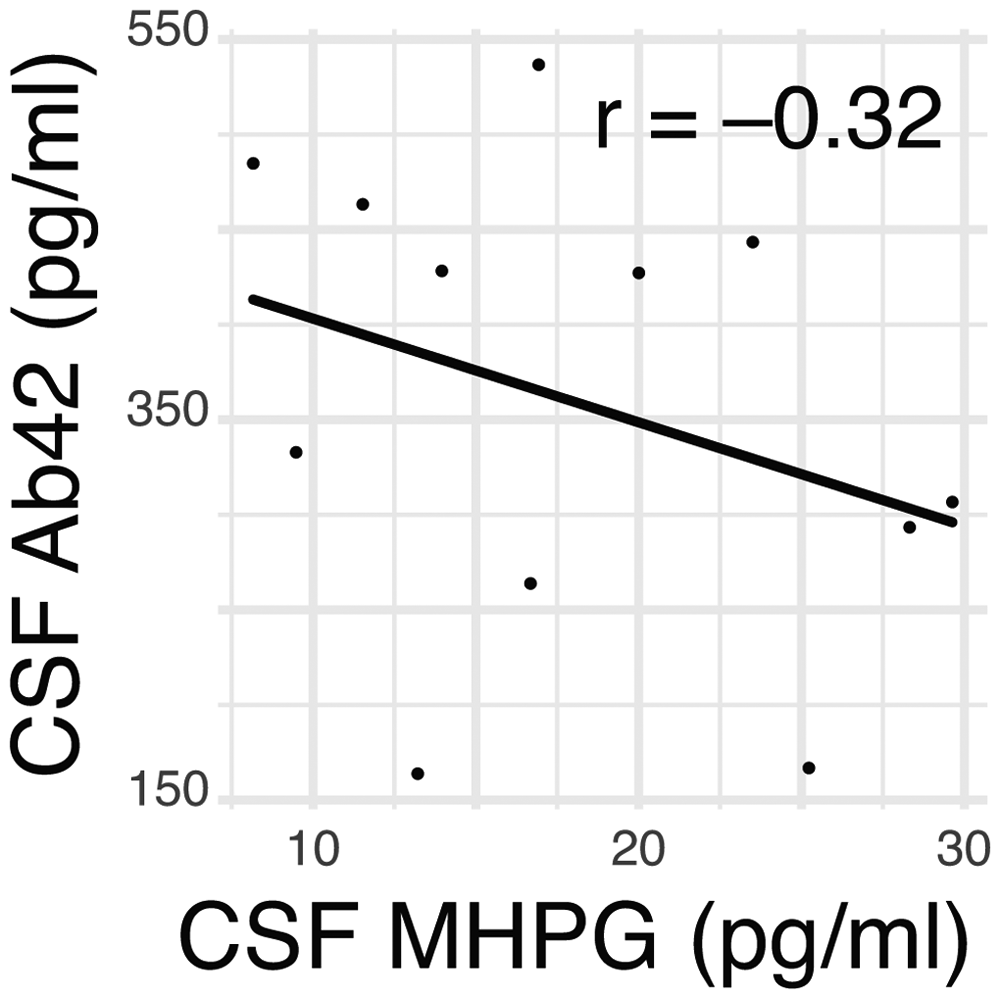
Exploratory data showing an inverse relationship between CSF Aβ42 and NE metabolite MHPG in PD.
It should be noted that the PD-FOG CSF biomarker pattern is distinct from that in AD. In PD-FOG, results demonstrated elevated Aβ42, reduced ptau, and fractalkine, whereas AD patterns show reduced Aβ42, elevated ptau, and fractalkine. This implicates Aβ42 as part of a distinct pathophysiology for FOG, perhaps related to the underlying dopaminergic loss seen in PD, and a potential therapeutic target.
Conclusion
Taken together, these findings suggest that NE, Aβ42, and inflammatory marker changes—interconnected in AD—may similarly interact in PD associated with the development of FOG, against a background of dopaminergic loss (Figure 6). We hypothesize that this pathophysiological loop could be critical for FOG development in PD, characteristically altered in FOG, and may predate its onset, justifying further study. One study has suggested that the onset of FOG in relation to this pathway may be mediated through the increases white matter hyperintensity load. 76 Recent reports published by an expert panel gathered by the International Society of Parkinson's disease and Movement Disorders Scientific issues committee indicate that data support a combination of dysfunctional circuits rather than a single one including the involvement of extranigral systems in PD-FOG.77,78 They recommended a systems biology strategy using a multifaceted approach for the study of biological markers that includes examination of multiple interacting systems in well characterized patients.
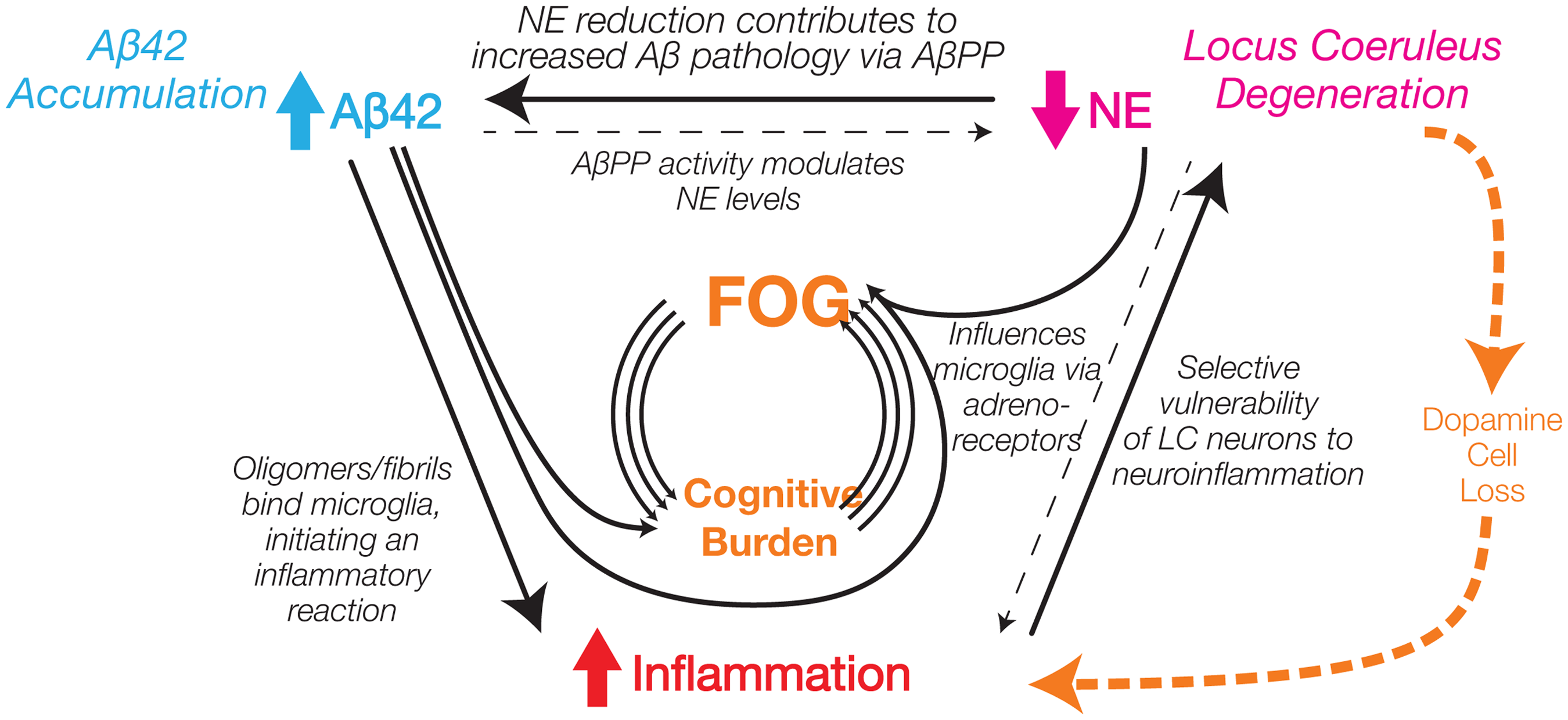
Schematic diagram illustrating interactions between non dopaminergic and dopaminergic systems hypothesized to contribute to FOG.
The examination of our hypothesis, that FOG results from a dysfunctional pathophysiological feedback loop incorporating NE depletion, amyloid accumulation, and neuroinflammation, that occurs concurrently with but distinct from primary PD dopaminergic pathology, fulfills such a requirement of multisystem approach. Future research should rigorously define and measure FOG with the levodopa challenge through objective modalities such as in a motion capture lab. It should include a multimodal study with deep characterization of chemical markers such as NE, Aβ42, and inflammatory markers through appropriate biological measures in CSF or serum and appropriate imaging such as amyloid or NET-PET. Finally, to discern whether such markers are predictive, a longitudinal design will be needed to assess conversion of those non-FOG patients to FOG, especially given the progressive degenerative nature of PD. A better understanding of the role of amyloid, NE and inflammatory markers in FOG and development of proper severity measures could result in rapid organization of trials to test existing amyloid clearing treatments and/or noradrenergic drugs in appropriate patients. Increasing NE transmission with atomoxetine has recently been shown to beneficially modulate CSF neuroinflammatory markers in mild cognitive impairment. 79 Carefully measuring FOG phenotypes will allow us to better select patients who might benefit from such treatments. For example, the NET-PET study demonstrated NE depletion particularly of OFF-FOG patients. We realize that Aβ42, NE, and inflammatory measures may not be the only markers and may possibly relate to a particular type of FOG. FOG is extremely complex because of its heterogeneity and may be the result of multiple system failures. Additional future studies could be modeled linking this feedback loop to other potential markers pointed to in the literature such as cholinergic, serotonergic and others. Dysfunction of such systems could act independently in particular phenotypes of FOG or in conjunction with this NE feedback loop and dopaminergic loss. This remains to be deciphered. And whether this pathophysiological loop has a role in the development of FOG in atypical parkinsonism has yet to be studied.
Footnotes
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the The McCamish Parkinson's Disease Innovation Program at Georgia Institute of Technology; The Curtis Family Fund; The Sartain Lanier Family Foundation; CS Foundation; Miracle for Mom Foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. However, they have the following disclosures:
Factor: Honoraria: Biogen, Takeda, Neurocrine. Grants: Medtronics, Boston Scientific, Sun Pharmaceuticals Advanced Research Company, Aspen, Biohaven, Neurocrine, Voyager, Prilenia Therapeutics, CHDI Foundation, Michael J. Fox Foundation, NIH 1 P50 NS123103-01, NIH 1R01NS125294-01, Parkinson Foundation. Royalties: Demos, Blackwell Futura, Springer for textbooks, Uptodate. Other Cronos
Weinshenker: Consultant CuraSen Therapeutics.
McKay: None