
Abstract
Parkinson's disease (PD) is traditionally recognized as a neurodegenerative disorder characterized by motor dysfunction and α-synuclein protein accumulation in the brain. However, recent research suggests that the circulatory system may also contribute to PD pathogenesis through the spread of α-synuclein beyond the brain. The blood-brain barrier (BBB), a key regulator of molecular exchange between the bloodstream and the brain, may become compromised in PD, allowing harmful substances, including pathogenic forms of α-synuclein, to infiltrate the brain and promote neurodegeneration. Transport mechanisms such as P-glycoprotein and the low-density lipoprotein (LDL) receptor-related protein (LRP-1) further modulate the movement of α-synuclein across the BBB, influencing disease progression. Additionally, extracellular vesicles are emerging as crucial mediators in the dissemination of α-synuclein between the brain and peripheral tissues, facilitating its spread and accumulation. The lymphatic system, responsible for clearing α-synuclein, may also contribute to PD pathology when impaired. This review highlights the growing evidence for a circulatory axis in the initiation and progression of PD. We propose that future research should explore the hypothesis that the circulatory system contributes to the pathogenesis of PD by aiding the distribution of α-synuclein throughout the body.
Plain language summary
Parkinson's disease (PD) is known for its effects on movement and the brain. But the cause remains unknown. Recent research suggests that problems with blood and blood flow contribute to the development of PD. For instance, proteins linked to PD have been identified in blood and heart tissues, indicating they could be early signs of the disease. Changes in heart rate, changes in blood pressure when standing up, and altered brain blood flow have been linked to PD. This review explores evidence that the circulatory system has important roles in the development and progression of PD. We propose further research to test if proteins in the blood start the disease and if blood vessels help spread it to the brain.
Keywords
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder primarily known for its impact on the motor system, though it also manifests with a broad spectrum of non-motor symptoms. 1 The pathological hallmark of PD is the accumulation of α-synuclein protein, which aggregates to form Lewy bodies and Lewy neurites in the brain. 1 Historically, PD was viewed as a disorder confined to the central nervous system (CNS), characterized by these intraneuronal α-synuclein deposits. Recent discoveries suggest that α-synuclein pathology extends beyond the CNS, implicating circulatory system involvement in the disease's progression.2–5 This is consistent with observations of structural and functional alterations in the circulatory system across a spectrum of neurodegenerative diseases, including Alzheimer's disease (AD), amyotrophic lateral sclerosis, Huntington's disease, and multiple sclerosis.6,7
The blood-brain barrier (BBB), a highly selective barrier that regulates molecular exchange between the bloodstream and the brain, may become compromised in PD. This disruption could facilitate the entry of harmful substances, including peripherally derived α-synuclein aggregates, thereby exacerbating neurodegeneration. 8 The BBB's integrity is maintained by complex interactions between endothelial cells, pericytes, microglia, and astrocytes.9,10 Disruptions in these interactions can increase BBB permeability, contributing to the neuroinflammatory environment observed in PD. 8 However, whether these changes are causal or reflect the CNS pathology remains to be determined. Despite this, the presence of α-synuclein in peripheral blood and its ability to cross the BBB via extracellular vesicles (EVs), which can transport α-synuclein across the BBB in both directions, may play a critical role in disseminating pathogenic proteins and disease progression11,12 thereby suggesting that bidirectional communication occurs between the CNS and the peripheral circulatory system. 12
Emerging evidence also supports a role for the lymphatic system in clearing α-synuclein from the CNS.13,14 Specifically, meningeal lymphatic vessels (MLVs), recently identified in rodents and humans, 15 facilitate the removal of waste products from the brain to the deep cervical lymph nodes (dCLN).13,14 Impairments in this drainage system may contribute to CNS α-synuclein accumulation, promoting aggregation and the subsequent development of PD pathology. 16
In this review, we discuss the connections between vascular changes, α-synuclein propagation, and PD pathology. While much of the evidence is correlative, we contend that understanding the causal link between BBB disruptions, blood-derived factors and α-synuclein propagation 8 in the pathogenesis of PD will provide pivotal insights for identifying new therapeutic targets.
Vascular changes in PD: a gateway to PD pathology
Vascular changes (i.e., alterations in the structure, function, or properties of blood vessels) are a common feature of AD, Huntington's disease, amyotrophic lateral sclerosis, multiple sclerosis, and PD.6,7 In some individuals with PD, disruption of the BBB has been identified as a vascular issue. 17 Describing and understanding the structures that constitute the BBB and their close interaction is key for understanding BBB dysfunction in PD development and identifying potential therapeutic strategies.
The BBB is a highly selective membrane (Figure 1B) that controls the transport of molecules (e.g., glucose and amino acids) between the circulatory system and the brain.18,19 BBB function depends on specific interactions between the endothelial, vascular smooth muscle, and pericyte cells. Together, these cells form part of the neurovascular unit (NVU) in close collaboration with neurons and different glial cells, including microglia, oligodendrocytes, and astrocytes (Figure 1A and B).9,10 The complex neurovascular network helps to maintain adequate nutrient delivery, a balanced neural environment, and healthy cerebral blood flow,9,20 while preventing the entry of undesirable compounds, pathogens, etc.
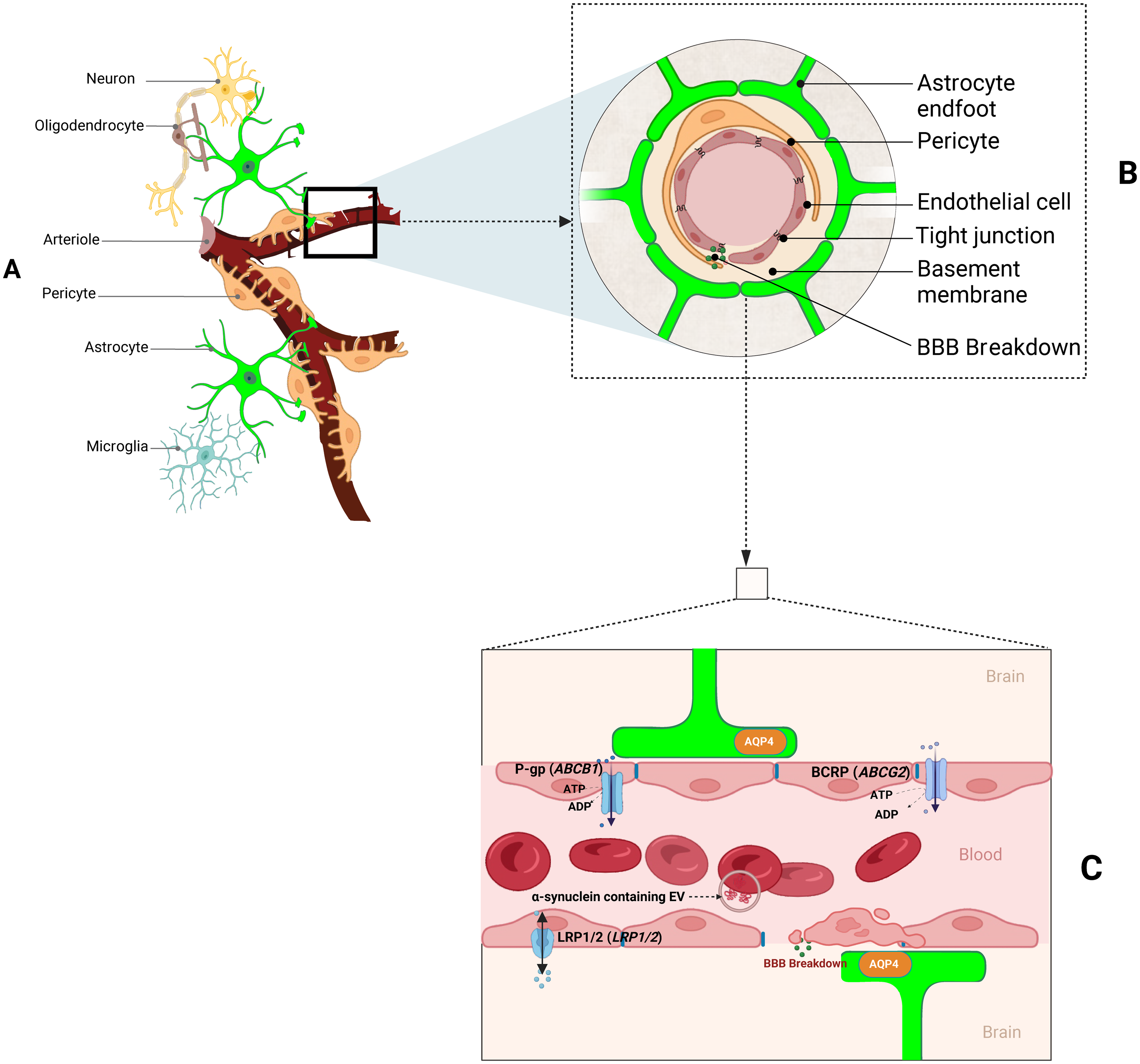
Cartoon illustrating the components and potential mechanisms through which the neurovascular unit (NVU) can dysfunction in PD. (A) The NVU includes complex cellular interactions between neurons, oligodendrocytes, pericytes, astrocytes, and microglia. (B) A cross-sectional diagram of the NVU highlighting the BBB. The BBB is made up of pericytes and endothelial cells joined by tight junctions, all encased in astrocytic end feet and bordered by a basement membrane. (C) A magnified view of a longitudinal section of the BBB. This panel illustrates the AQP4 mainly localized to the perivascular astrocytic end feet and the presence of various carrier pumps on the endothelial membrane.
As a result of the close interactions that occur between the NVU cells, disruption of the molecular processes in any of them may compromise the overall integrity of the BBB through a domino effect that reduces selectivity and potentially initiates pathways associated with neurodegeneration. 10 For example, microglial activation triggers the release of pro-inflammatory molecules (e.g., cytokines, such as IFN-γ, TNF, and IL-12), thereby inducing the A1 subtype of reactive astrocytes, which in turn makes the BBB hyperpermeable and contributes to neuroinflammation.21–24 Similarly, astrocytes25–28 and brain pericytes,29–31 when stimulated by classically activated neuroinflammatory microglia, can also release pro-inflammatory and angiogenic (e.g., vascular endothelial growth factor (VEGF)) molecules that increase inflammation and stimulate the development of immature blood vessels and thus compromise the BBB. 8
Transport carriers, such as P-glycoprotein (P-gp) and low-density lipoprotein receptor-related protein-1 (LRP-1), are found in the membranes of the endothelial cells that make up the BBB. These carriers are also present in the apical and luminal membranes of capillary endothelial cells (Figure 1C).32,33 These transporters have vital roles in regulating the influx and efflux of molecules between the blood and brain.10,34 For example, the P-gp efflux pump is expressed on the luminal surface of the BBB's endothelial cells and is actively involved in the cellular detoxification and efflux of a wide range of molecules (e.g., drugs and toxins) from the brain. 35 However, the function of P-gp decreases with age and is particularly impaired in the midbrain of individuals with PD.9,36 Reductions in P-gp activity may indirectly increase BBB permeability while simultaneously reducing the endothelial cells’ ability to remove these harmful substances from the brain. This was demonstrated by Kortekaas et al. (2005), who identified an increased uptake of the radiolabelled P-gp substrate [11C]-verapamil into the midbrain of PD patients, compared to healthy controls. 37 Reduced P-gp function is a late occurrence in PD that may accelerate ongoing neurodegeneration.36,38 Notably, blocking P-gp in animal models of PD increases brain permeability to FLZ, formulated as N-2-[(4-hydroxyphenyl)-ethyl]-2-(2,5-dimethoxy-phenyl)-3-(3-methoxy-4-hydroxyphenyl)-acrylamide, a synthetic squamosamide derivative that has potent neuroprotective effects with potential anti-PD properties.39–44 FLZ has been approved for PD treatment by the Chinese National Medical Products Administration and is undergoing Phase I clinical trials in China. 43 Therefore, it remains possible that inhibiting transport carriers such as P-gp may be a therapeutic strategy for reducing the efflux and therefore increasing the net uptake of anti-PD drugs to the brain. However, the potential benefits of increased drug delivery (e.g., FLZ) need to be carefully weighed against the possibility that P-gp inhibition might also exacerbate disease progression by enabling the increased accumulation of toxins. Therefore, further investigations are necessary to determine the optimal approach for P-gp modulation in PD treatment.
In addition to P-gp, other transporters (e.g., LRP-1) may contribute to alterations in BBB permeability in PD. LRP-1 is a bidirectional transporter that mediates the influx and efflux of lipoproteins (e.g., ApoE, APOA4) and other molecules (e.g., Cytochrome C, Albumin, Ca2+) across the BBB.12,45–47 A radioactive α-synuclein tracer in mice demonstrated that LRP-1 facilitates the transport of α-synuclein from the blood to the brain and vice versa. 12 LRP-1 controls the neuronal uptake and spread of α-synuclein, particularly monomers and oligomers. 48 Long-term reductions of α-synuclein distribution in neurons are caused by a decrease in LRP-1 expression. 48 Peng et al. (2022) observed elevated levels of LRP-1 in PC12 immortalized sympathetic neuron model cell line transfected with both the full-length and N-terminus of α-synuclein. These observations were consistent with a feed-forward loop in which α-synuclein drives LRP-1 expression, potentially accelerating the spread of pathogenic α-synuclein. Reducing LRP-1 expression in mice significantly inhibited the spread of α-synuclein between neurons. 49 Notably, changes in ApoE and LRP-1 expression and the lipoprotein balance of melanised neurons in the substantia nigra occur early in the development of PD. These changes are thought to be part of a protective mechanism to reduce harmful α-synuclein aggregation 50 and support the hypothesis that decreasing LRP-1 expression is a potential therapeutic target for early PD intervention. 50
Variations in BBB integrity characterize PD
The evidence regarding BBB dysfunction in PD is inconsistent.17,51,52 For example, magnetic resonance imaging (MRI) has identified increased BBB leakage of the gadolinium contrast agent in PD patients’ basal ganglia compared to healthy controls. 17 However, the observed changes were subtle, not universal across brain regions, and resembled those in cerebrovascular disease. 17 In contrast, Fujita et al. (2021) observed no evidence of BBB leakage in PD using PET with a different tracer. 53 This apparent contradiction might be due to different methodological sensitivities (MRI vs. PET). Finally, imaging studies have shown increased permeability and leakiness of the BBB in the substantia nigra and post-commissural putamen in people with PD.54,55 This further highlights the heterogeneous nature of the progression of the disease.
A recent study using machine learning and deep learning to analyse phenotypic progression identified three distinct subtypes of PD based on progression rate: 1) Inching Pace subtype (PD-I), with mild baseline severity and slow progression, 2) Moderate Pace subtype (PD-M), with mild baseline severity and moderate progression, and 3) Rapid Pace subtype (PD-R), marked by the fastest symptom progression. 56 Transcriptomic profiles of these subtypes suggest that neuroinflammation, oxidative stress, metabolism, PI3K/AKT, and angiogenesis pathways drive the rapid progression in the PD-R subtype (CNTNAP1, TRIM40, RETREG3, FYN, MBNL2, ATP6V0A1, STAT3, BECN1, BAG6, HLA-DRB1, HLA-DRB5, HLA-DQB1, SIPA1L2, and PRRC2A). 56 Given the shared genes between the transcriptomic-inferred pathways of the PD-R subtype and those associated with pathological brain vascular endothelial cells, 57 investigating BBB function in these subtypes could provide valuable insights into BBB susceptibility and PD progression.
The role of blood α-synuclein and EVs in PD
α-synuclein is highly expressed within whole blood, 58 with high levels of phosphorylated α-synuclein detected in the plasma of individuals with PD.59–62 Red blood cells (RBCs) account for the greatest proportion (>99%) of α-synuclein within the blood, 63 while mononuclear cells (0.05%) and platelets (0.2%) account for lower levels. 63 However, accounting for absolute cell numbers, platelets contain the highest per-cell concentrations of α-synuclein. 63 There is growing interest in quantifying α-synuclein from blood as a biomarker for early disease detection, monitoring disease progression, and assessing treatment responses. To this end, an assay has been developed to detect neuronally derived extracellular vesicle α-synuclein in the blood. 64 The utility of blood α-synuclein assays is due to the ease of accessibility and non-invasive nature of sample collection.
It is well-established that α-synuclein misfolds and aggregates into toxic fibrils in PD. 65 A portion of circulating α-synuclein is encapsulated in exosomes. 66 Exosomes are nanometre-sized extracellular vesicles that originate from multivesicular bodies within cells and play a role in cell-to-cell communication. 67 These exosomes encapsulate various molecules, including proteins, within a lipid membrane, which are then transported to the cellular membrane and secreted to recipient cells. 67 Notably, the microenvironment within exosomes promotes the misfolding and aggregation of α-synuclein, suggesting that exosomes could contribute to pathogenic α-synuclein transmission. 68 Additionally, exosomes have been found to enhance the uptake of oligomeric α-synuclein by recipient cells compared to its free form through receptor-mediated endocytosis and phagocytosis. This indicates that exosomes may play a more significant role in the disease process than their low overall abundance of total extracellular α-synuclein would suggest. 69
RBCs have been observed to release α-synuclein containing EVs capable of crossing the BBB, 11 resulting in oligomeric α-synuclein deposition within astrocytes and microglia.11,70 It is possible that RBC-derived EVs circulating in the bloodstream are interacting with the BBB, gut, and other tissues in response to inflammation. 71 Recent findings demonstrate that RBC-EVs readily migrate to the gut, disrupting the gut-vascular barrier and acting as a primary α-synuclein source. 72 This α-synuclein could then be transported to the brain via the vagal nerve.73–76 Future research should empirically determine the mechanisms facilitating RBC-EV α-synuclein transfer to neural structures and whether this is a primary cause or secondary effect of PD.
Individuals with PD have heightened inflammatory responses, characterized by elevated levels of inflammatory cytokines triggered by α-synuclein-containing RBC-EVs in blood immune cells and increased sensitivity of monocytes. 77 This is consistent with the presence of peripheral inflammation in PD.78–80 and suggests a systemic peripheral inflammatory state that may contribute to the disease's progression. This inflammatory hyperactivation is associated with increased LRRK2 production and kinase activity.77,81 Notably, inflammatory sensitization is observed when THP-1 cells, a human leukaemia monocytic cell line, are treated with RBC-EVs isolated from individuals with PD. 77
Platelet activation contributes to blood α-synuclein propagation and subsequent PD pathogenesis.82,83 A potential causal link between elevated platelet distribution width (PDW), a marker of platelet activation, and PD development has been identified.84,85 Individuals with PD have heightened blood clotting tendencies, which is associated with increased platelet activation. 86 Considering the substantial abundance of α-synuclein in blood platelets, its presence within platelet-derived EVs, 87 and the fact that PD-EVs represent the most abundant EV population in blood plasma, 88 platelets may contribute to the dissemination of α-synuclein throughout the body.
The role of the lymphatic system in α-synuclein propagation: implications for PD
Meningeal lymphatic vessels (MLVs), or lymphatic vessels in the dura mater of mice, were identified in 2015. 14 MLVs have been visualised in non-human primates and humans, 15 where they mediate the flow of soluble components between the interstitial fluid (ISF) and cerebrospinal fluid (CSF).13,14 Additionally, MLVs facilitate the passage of immune cells, misfolded proteins, cellular debris, and metabolites into the deep cervical lymph nodes (dCLNs) from the CSF, which serve as a conduit between the brain and peripheral circulation.13,14
The CNS depends on MLV function to maintain homeostasis, and MLV draining function declines with advancing age. 89 Since old age and the accumulation and misfolding of α-synuclein protein are key drivers of PD, the elimination of α-synuclein is crucial for PD treatment. 90 Impairment of MLVs draining function affects the development of PD. 16 Ligation of the deep dCLNs in A53T mice was associated with an increase in α-synuclein pathology when compared to control mice, however, whether this is sufficient to cause PD remains to be determined. The cervical lymphatic ligation that caused this disturbance in meningeal lymphatic outflow increased inflammatory cytokine production and reactive gliosis in the CNS. 16 Notably, reduced drainage from MLVs to the dCLNs as well as cognitive and motor impairments were seen in a mouse model of PD that had preformed α-synuclein fibrils injected into its brain. 91 Data from human patients validates the involvement of MLVs in the aetiology of PD. The flow of MLV flow was assessed in 319 patients with atypical parkinsonian disorders 375 patients with idiopathic PD and 371 non-affected control individuals using dynamic contrast-enhanced (DCE)-MRI. CSF flow via the MLVs and perfusion to the dCLNs were found to be lower in idiopathic PD patients than was measured in the other two groups. 92 Meningeal inflammation and meningeal lymphatic endothelial barrier failure were also brought on by MLV dysfunction. 92 Thus, meningeal lymphatic drainage is distinctively impaired in idiopathic PD.
The glymphatic system has a crucial role in clearing waste products from the CNS, particularly during sleep. 93 Specifically, the glymphatic system contributes to brain-cleaning by promoting the flow of ISF into the lymphatic system and the influx of CSF into the brain parenchyma (Figure 2). One of the most prevalent early signs of neurodegenerative diseases, such as AD and PD, is sleep disturbance. 94 It is possible that the neuroprotective effects of sleep may be mediated through the glymphatic system. 93 LRRK2 R1441G disrupts glymphatic function by directly phosphorylating and depolarizing AQP4, which is necessary for the proper functioning of the brain's glymphatic system (Figure 2). This impairment leads to IFNγ accumulation, exacerbating existing neuroinflammation and neurotoxicity. 95 Notably, LRRK2 inhibition can mitigate these detrimental effects, highlighting a role for the glymphatic system in LRRK2-associated neuroinflammation. 95 This also introduces the possibility that inherited changes in LRRK2 can make a causal contribution to PD by disrupting glymphatic function.
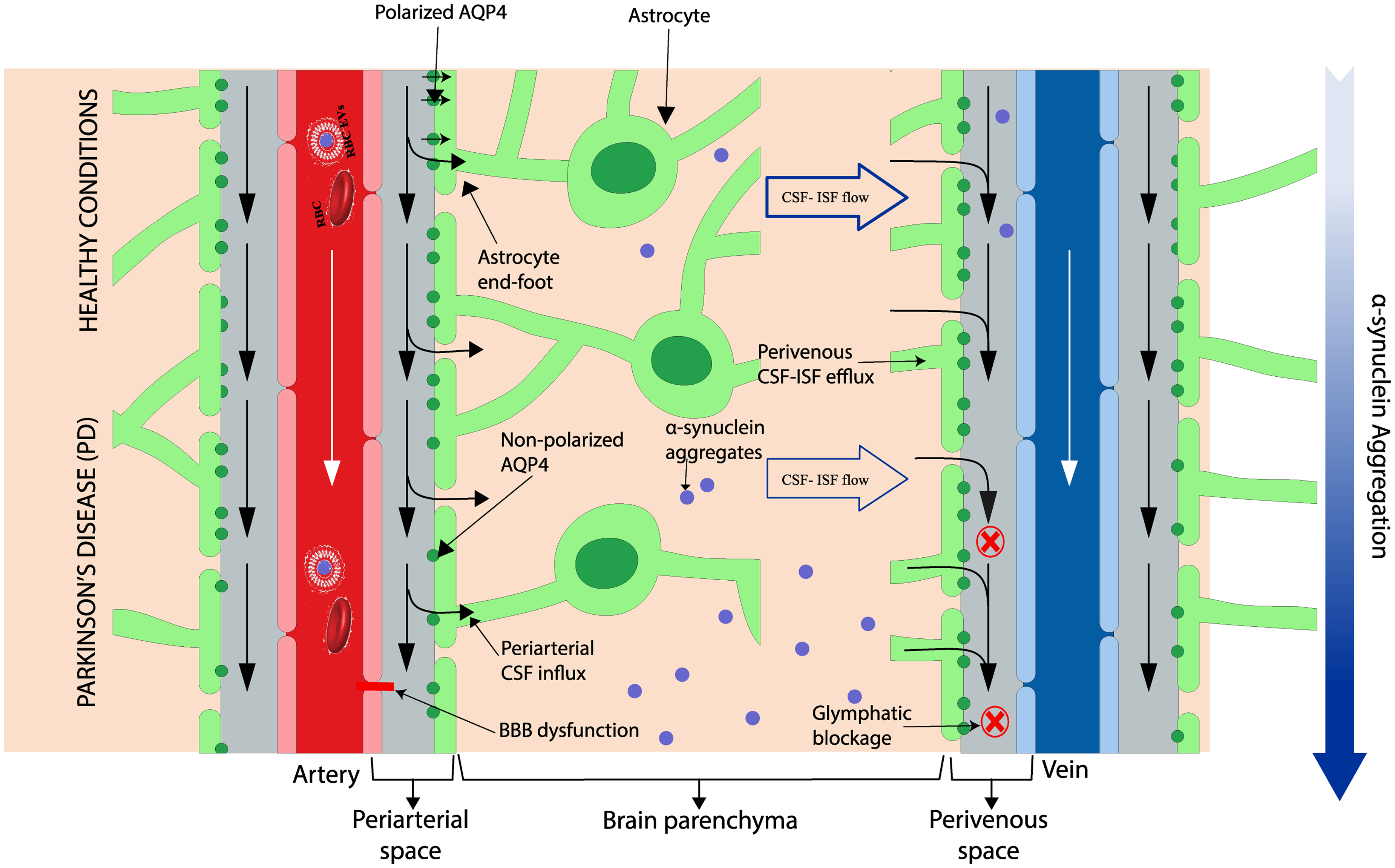
Glymphatic system in healthy vs PD condition. In healthy conditions, the glymphatic system facilitates the clearance of α-sb ynuclein by promoting the flow of cerebrospinal fluid (CSF) through astrocyte end-feet. However, in pathological conditions such as PD, BBB dysfunction, disruptions in the glymphatic system, including loss of AQP4 polarization and impaired CSF-ISF flow, contribute to α-synuclein accumulation. Red “X” indicating the blocked or impaired clearance pathways in PD, highlighting glymphatic dysfunction. The white arrows (downward) represent the normal blood flow through the arteries and veins in healthy conditions and in PD. The black arrows indicate fluid transport along the periarterial space into the brain parenchyma and out along the perivenous space. Image modified from Ray et al. (2019). 96
Perivascular space (PVS) are fluid-filled regions surrounding tiny blood vessels in the brain, and their enlargement was previously observed in AD. 6 The integrity of the PVS can be compromised by factors that result in its expansion. These factors include, but are not limited to, BBB breakdown, pericyte deficiency, astrocytic dysfunction, and decreased CSF drainage. 97 Donahue et al. (2021) combined MRI with a novel image processing technique to visualize and measure PVS volume fractions in the brains of 179 idiopathic PD patients, 67 familial PD patients, 101 non-manifest carriers of PD-associated mutations, and 84 healthy controls. 97 Compared to non-manifest carriers and controls, global PVS volume percentages were significantly greater in the idiopathic PD and familial PD groups. 97 Similarly, patients with LRRK2-associated PD exhibit enlarged PVS, consistent with a role for LRRK2 in glymphatic system dysfunction. 92 PVS enlargement in PD patients is particularly noticeable in the medial orbitofrontal and superior temporal regions, 92 areas associated with cognitive functions often impaired in PD.98,99 Collectively, the evidence indicates that changes in MLV and PVS may function as biomarkers for PD progression and provide an understanding of disease heterogeneity and the role of the circulatory system in the propagation of α-synuclein throughout the development of PD. Targeted treatment strategies may result from further investigation into the molecular mechanisms behind PVS expansion, such as disruption of the BBB and aberrant immunological responses.
Peripheral α-synuclein accumulation and neural propagation
Previous α-synuclein propagation hypotheses have focused on the neural spread100,101 in attempts to explain observations of Lewy bodies in the brain and spinal cord, sympathetic trunk ganglia, cardiac nerve cells, and other peripheral autonomic networks in PD patients.102,103 Braak's highly influential hypothesis posits that synucleinopathy lesions originate in the peripheral nervous system and spread through the autonomic nerves to the dorsal motor nucleus of the vagus nerve, and ultimately end up in the cerebral cortex. 104 Several experiments support predictions from this hypothesis. For example, when α-synuclein fibrils were injected into the duodenum and pyloric muscularis layers, areas with significant vagus nerve innervation, pathological α-synuclein lesions subsequently appeared in the brains of the test mice, with initial changes observed in the dorsal motor nucleus of the hindbrain. 76 Furthermore, vagotomy, which involves severing the autonomic nerve pathway, almost completely blocks the spread of α-synuclein from the gastrointestinal tract to the brain. 76 High concentrations of pathological α-synuclein have been observed in the appendix, although there is contradictory evidence that appendectomy delays PD onset.105,106 However, neuropathological evidence implicates α-synuclein pathology in the brain and not the other tissues. 107 This raises the possibility that peripheral α-synuclein deposits may result from leakage from the CNS rather than a de novo origin.
The brain-first vs body-first hypothesis for PD was put forth by Borghammer et al. (2019). According to this concept, the autonomic nervous system is eventually affected by the initial α-synuclein pathology that occurs within the CNS, most likely above the substantia nigra pars compacta. 101 When PD occurs in a body-first manner, the pathogenic α-synuclein starts in the PNS and enteric nervous system and moves caudo-rostrally to the autonomic and CNS. These subtypes have been validated using multimodal imaging methods, with brain-first PD showing sequential pathology in the amygdala, substantia nigra, locus coeruleus, dorsal motor nucleus, and heart, while body-first PD shows sequential pathology in the intestine, heart, dorsal motor nucleus, locus coeruleus, and substantia nigra. 108 Despite these insights, the mechanisms underlying the propagation of α-synuclein throughout the body are still under investigation, particularly the role of non-neural pathways in the spread of α-synuclein pathology. Notably, as for all potential mechanisms contributing to α-synuclein pathology and PD development, these mechanisms need not be mutually exclusive and may act in combination. 109
Peripheral α-synuclein accumulation and systemic propagation
The presence of pathological α-synuclein in the blood and CSF has been validated, and both have been linked to PD symptoms and disease progression.11,110 Beyond the blood and CSF, pathological α-synuclein has also been found in other bodily fluids, including saliva and lymph.111,112 For example, plasma α-synuclein levels have been reported to correlate with specific PD symptoms. 113 Similar to neural pathways, the circulation of bodily fluids—such as plasma, CSF, ISF, and lymph—facilitates the transmission of pathological α-synuclein between the brain and peripheral organs.92,93
In line with the accumulation of α-synuclein in peripheral tissues in PD, a recent report has suggested a potential role of the liver in the disease's progression. 114 For the first time, this study demonstrated that human hepatocytes can uptake α-synuclein assemblies in vitro. 114 Reyes et al. (2021) found that human postmortem liver tissue from individuals with neuropathologically confirmed α-synuclein pathology showed α-synuclein presence within hepatocellular structures at a significantly higher rate (75%) compared to control subjects without brain α-synuclein accumulation (57%). 114 Although the role of α-synuclein deposition in the liver in both humans and mice remains unclear, it is hypothesized that α-synuclein might be transported from either the gut or brain to the liver via nerve connections or blood vessels, where it could be cleared as part of the liver's detoxification process. 114 Given the liver's high density of nerves and blood vessels, 115 both scenarios are plausible, but further research is needed to clarify the exact mechanisms.
Notably, human hepatocytes have been shown to reduce extracellular levels of α-synuclein over time through internalization, partly mediated by the gap junction protein connexin-32, 114 which opens new avenues for reconsidering liver damage in the context of α-synuclein deposition and its connection to PD pathogenesis. Liver disease may cause metabolic waste products and toxic compounds to accumulate in the ISF of the brain, which could lead to neuronal malfunction and cognitive decline. 116 This was verified in a rat model of chronic liver disease created by ligating the bile duct. The rat model showed decreased AQP4 and altered glymphatic clearance in multiple brain regions, including the hippocampus, prefrontal cortex, and olfactory bulb. These outcomes correspond with deficiencies in cognition and behaviour. 116
A recent study looked for temporal connections between liver insult and Parkinsonian symptoms in the medical records of patients with PD and chronic liver impairment brought on by cirrhosis or liver metastases. 117 35 PD patients who had either liver metastases or cirrhosis and chronic liver damage were included in the study. For each of the 22 patients with PD with liver metastases, the cancer diagnosis came first. On the other hand, liver dysfunction was frequently identified in patients with cirrhosis before PD. There was no explanation for this disparity in age at diagnosis. This data supports the hypothesis that PD and cirrhosis may be clinically related. 117
Exploring the mechanisms of α-synuclein dissemination in PD
The heterogeneous nature of PD is highlighted by the number of different current theories on the propagation of α-synuclein, many of which fail to consider the involvement of the circulatory system.4,108 Traditionally, a neurological perspective has been used to explain the spread of pathogenic α-synuclein. However, recent discoveries highlight the complexity of PD pathogenesis by indicating the participation of both neural and circulatory 92 mechanisms, as well as bidirectional in some cases, in the dispersion of α-synuclein.
To address the limitations of existing models, we propose a bidirectional propagation model for α-synuclein involving both neural and circulatory pathways (Figure 3). The neural propagation hypothesis suggests that α-synuclein spreads from the brain to other peripheral structures, such as the enteric nervous system, heart and liver or from these peripheral tissues to the brain through established neuroanatomical connections and autonomic circuits.118–120 Although the exact mechanisms of this neural propagation are not yet fully understood, several pieces of evidence support this hypothesis. For instance, Lewy bodies in the atrial ganglia, coronary artery nerve fibres, and myocardium of PD patients support the idea of neural propagation.114,121 However, it remains possible that the observed α-synuclein aggregates in cardiac tissues could also be formed de novo rather than solely spreading from the nervous system.
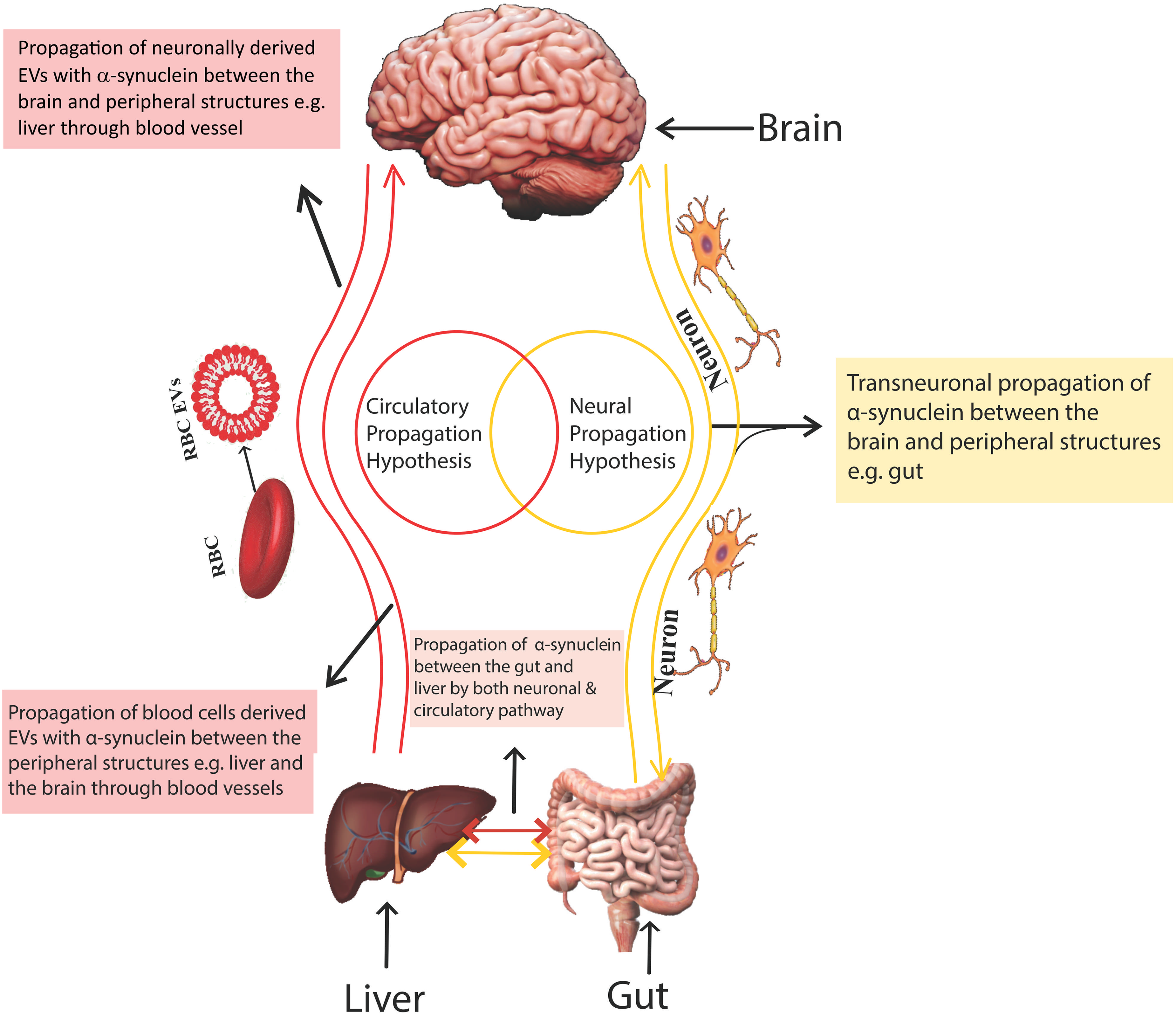
α-Synuclein distribution hypotheses. This figure illustrates two primary hypotheses regarding the propagation of α-synuclein in the body, which is implicated in the pathogenesis of PD.
The circulatory hypothesis proposes that pathological α-synuclein originates from brain leakage across the BBB and blood-CSF circulation, which communicate with the autonomic nervous system. Alternatively, α-synuclein could form de novo in peripheral tissues. The detection of α-synuclein in body fluids and its accumulation when the lymphatic system is impaired supports this hypothesis, making it a compelling avenue for further investigation.16,111,112,114
Peripheral organs that are heavily vascularized and innervated, including the liver and heart, display malfunction associated with an accumulation of α-synuclein, which may pave the way for PD. For example, the liver may be involved with the clearance of abnormal α-synuclein. 114 Studies have demonstrated that overexpression of α-synuclein in the perivascular nerve fibres lessens the contraction of the mouse aorta caused by norepinephrine, suggesting a possible effect on vascular function. 122 Reyes et al. (2021) showed that α-synuclein was transported from the brain to the liver after a single stereotactic injection of protein oligomers into the striatum of wild-type mice. One month after the injection, protein buildup was observed in the liver. 114 Although the precise pathway of distribution from the brain to the liver is still unknown in humans and animals, nodosa ganglia efferent neurons, which link the hepatic arteries and portal veins, are thought to play a role in this process. 114 Furthermore, the liver is directly innervated by branches of the splanchnic and vagal nerves through the portal area, which is connected to the bile ducts and portal vein. 115 These pathways offer logical paths for α-synuclein transport to the liver, whether directly from the brain or through the hepatic arteries and portal veins from the gut. Alternatively, it is not impossible that α-synuclein may develop in the liver by de novo means. The observed association between PD and liver cirrhosis 117 lends support to the hypothesis that liver dysfunction may contribute to α-synuclein accumulation and PD progression.
The neural propagation and circulatory hypotheses suggest that pathogenic α-synuclein is propagated and spreads from a ‘central’ point. However, α-synuclein deposits may also form de novo due to tissue-specific expression or the localized release of high concentrations from circulating blood cells. Further research is needed to elucidate the role(s) of neural propagation and circulatory mechanisms in PD risk, disease development and progression.
Summary, research gaps, and future directions
In this review, we have explored the complex interplay between the circulatory and nervous system in PD. We highlighted vascular dysfunction, the close interactions within the NVU, and how disruptions can potentially lead to BBB dysfunction, neuroinflammation, and ultimately contribute to α-synuclein propagation and PD pathogenesis. We have discussed how alterations in blood-borne α-synuclein and the involvement of the lymphatic system can contribute to α-synuclein spreading from peripheral tissues to the CNS. Several variables, including EVs, inflammatory reactions, and endothelium transporters, are important in controlling how the brain and circulatory system interact, which can affect how PD progresses. The possibility of an intricate relationship between PD pathology and the circulatory system exposes knowledge gaps and research directions for the future concerning the development and risk of PD.
Firstly, although evidence suggests that BBB dysfunction contributes to PD, discrepancies in findings regarding BBB integrity need to be resolved.17,53 More studies are required to identify specific mechanisms that lead to BBB impairment in PD and to determine whether these changes are consistent across different stages or subtypes of the disease. For example, detailed meta-analyses are required to address this research gap and enable discrimination between the potential mechanisms that explain the divergent observations. Additionally, these investigations should be designed to incorporate the temporal dynamics of BBB maintenance and transporters (e.g., P-gp and LRP-1), focusing on different stages of PD and potential subtype-specific variations. Future studies should strive to clarify conflicting evidence regarding BBB leakage in PD, possibly due to methodological differences in sensitivity or study populations. They should also address the apparent contradictions between findings from animal models and human disease by concentrating on data from clinical studies whenever possible.
Secondly, the mechanisms by which EVs contribute to the propagation of α-synuclein within the CNS and between the CNS and peripheral tissues are not fully understood. Research is needed to clarify EV-mediated α-synuclein transport pathways and explore the impact of different EV populations on PD pathology. The use of innovative in vitro techniques (e.g., organ-on-chip platforms, human-derived cells, organoids, and patient-specific induced pluripotent stem cells) will enable the exploration of the complex interactions between the EVs and PD.43,123–126
Thirdly, the exact origins and pathways of α-synuclein propagation remain unclear. Although the brain-first versus body-first hypothesis provides a framework, more evidence is required to delineate how α-synuclein accumulation in peripheral organs (e.g., the liver or gut) impacts its spread to the brain. While the bidirectional spread of α-synuclein between the brain and gut has been extensively studied, confirmation of its spread from the gut to the liver in PD patients without brain α-synuclein deposition is required. 108 Further studies are necessary to elucidate the consequences of α-synuclein accumulation in the liver.
Fourthly, the role of the MLVs and the glymphatic system in clearing α-synuclein and other waste products from the brain is an emerging area.91,92 Clinical studies should focus on understanding how lymphatic dysfunction contributes to PD progression and whether enhancing lymphatic clearance could serve as a therapeutic strategy.
In conclusion, the involvement of the circulatory system in PD pathogenesis underscores the complexity of the disease and the multifaceted nature of α-synuclein propagation. Vascular dysfunction, BBB permeability, and the roles of peripheral organs and the lymphatic system contribute to the spread of α-synuclein, highlighting the need for a holistic approach to understanding and treating PD. Future studies can shed light on the involvement of the circulatory system in PD and ultimately develop more effective diagnostic and treatment approaches by addressing the research gaps mentioned above.
Footnotes
Acknowledgments
We gratefully acknowledge the generous donations from the Dines Family Trust.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: OO is the recipient of a University of Auckland scholarship. SF, AC and JOS were supported by an MJFF grant (ID 021131) to JOS and AC. SF was supported by a grant from the Dines Family Foundation to JOS.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. (or text from authors)