
Abstract
PTEN-induced kinase 1 (PINK1)-related Parkinson's disease (PD) is traditionally considered a rare autosomal recessive form of early-onset PD (EOPD), lacking classical Lewy body pathology. However, this characterization underestimates and oversimplifies PINK1-PD, largely due to a lack of extensive studies in diverse ethnic populations. This review and meta-analysis explores considerable variations in PINK1 variant rates and the wide heterogeneity influenced by patient- and variant-specific factors, delineating a more precise disease profile. Our findings reveal that PINK1-PD is more common than previously thought, with geographic ‘hotspots’ where up to 9% of EOPD cases are linked to PINK1 variants, including the pathogenic p.Leu347Pro variant affecting 1 in 1300 West Polynesians. Homozygous PINK1-PD typically manifests around age 35, predominantly affecting the lower limbs, with an excellent response to levodopa. Heterozygous PINK1-PD presents an ‘intermediate’ phenotype, with a later onset age (around 43 years) than homozygous PINK1-PD but earlier than idiopathic PD (typically after age 65). The severity of the phenotype is influenced by variant zygosity and pathogenicity, interacting with genetic and environmental factors to push some individuals beyond the disease threshold. Notably, females with PINK1-PD have earlier onset age than males, particularly in homozygous cases and when variants occur in the first half of PINK1's kinase domain. Contrary to traditional views, α-synuclein pathology is present in 87.5% of PINK1-PD postmortem cases across ages and variants. We challenge conventional views on PINK1-PD, highlighting distinct phenotypes influenced by zygosity, sex, and a role for α-synuclein pathology, urging for increased recognition and research of this not-so-rare disease.
Plain language summary
Variants in the gene PTEN-induced kinase 1 (PINK1) cause early onset Parkinson's disease, a form traditionally thought to be rare and lacking usual brain changes seen in typical Parkinson's. However, this may underestimate and oversimplify the condition, especially for certain ethnic groups. We analyzed 120 studies of 743 patients with PINK1-related Parkinson's, finding that in some areas, up to 9% of early-onset Parkinson's is due to PINK1 variants. One specific damaging variant, p.Leu347Pro, affects about 1 in 1300 West Polynesians, exceeding the threshold for a rare disease. People with PINK1 Parkinson's experience symptoms around age 35, mostly affecting their legs, and they respond well to the medication levodopa. Although it was thought two copies of a PINK1 variant were required to develop disease, those with one copy show similar Parkinson's symptoms but with a later onset, around age 43 (still earlier than typical Parkinson's which generally starts after age 65). Women with PINK1 Parkinson's tend to show symptoms earlier than men. This difference is more pronounced in those with two copies of the variant and when the variant occurs in a specific part of the PINK1 gene. Post-mortem studies reveal that a protein called α-synuclein, usually associated with typical Parkinson's, is present in 87.5% of PINK1 Parkinson's cases, highlighting it's involvement in this form of disease. Our study challenges the conventional understanding of PINK1 Parkinson's, describing unique characteristics dependent on the variant type. Given its higher rates in some regions, we call for greater recognition and research into PINK1-linked Parkinson's disease.
Keywords
Introduction
While classic Parkinson's disease (PD) is primarily an affliction of the aged population, characterized by bradykinesia, tremor, rigidity, and postural instability, individuals with early onset PD (EOPD) are symptomatic before 50 years of age and can carry these devastating effects with them for the majority of their lives.1,2 EOPD comprises approximately 10% of 8.5 million individuals worldwide living with PD,3,4 and the second most common cause is variants in the gene PTEN-induced kinase 1 (PINK1).2,5 Some similarities emerge across previously reported patients with PINK1 variants. Typically, individuals present around 30–40 years of age with PD symptoms predominantly in the lower limbs, an excellent prolonged response to levodopa pharmacotherapy (though often developing drug-related dyskinesia) and slow disease progression.6–9 This characteristic phenotype bears striking dissimilarities to typical late onset, rapidly deteriorating, idiopathic PD.
In the present review and meta-analysis, we performed a systematic search of the current literature on PINK1 variants in PD to investigate the specific factors at play in this under-researched disease. While most reports quote PINK1 PD as a rare homozygous recessive disease, the fluctuation in these observed rates suggests regional variability, with the population rates of PINK1 variants ranging between 1–9% of all EOPD cases.2,6,9–11 We debate the role that zygosity, ethnicity, sex and other genetic and environmental factors play in manifesting this typical phenotype and age of onset, positing that it is more beneficial to consider the distinct profile of PINK1 PD as a separate spectrum of disease. Lastly, we explore the association between PINK1 PD and α-synuclein (α-Syn) pathology.
Methods
Our meta-analysis included 120 studies that met our criteria. A full description of the methodology is available in Supplemental Information 1. We performed a systematic search using PubMed, Scopus, Web of Science (All Databases), and Google Scholar databases for relevant articles published before 31 August 2023. We used the search terms “Parkinson's disease”, “PINK1” and one of “Europe*”, “Asia*”, “Africa*”, “Latin*” or “Polynesia*”. The total number of records returned was 415, from which 145 duplicates were removed. The remaining 270 records were screened to exclude 177 studies written in a language other than English or if they were not primary articles studying human PINK1 PD patients (investigative studies screening for PINK1 variants in patients (even if not detecting any present) were included). The remaining 93 reports were all retrieved and subsequently assessed to determine if they classified the relevant patient ethnicity, deriving 75 included articles.5,12–85 An additional 45 studies were included outside of the systematic search, as they either investigated multiple ethnicities across the described categories or were found separately throughout the literature scope (a portion through the Human Gene Mutation Database®).6,9,11,86–127 These studies met the criteria for inclusion and thus were included in the following analysis.
All variants were reported unless they were described as a benign common polymorphism. For each individual, available patient PINK1 variant information was recorded (specific variant, zygosity, reported pathogenicity), as well as their available demographic information (age at disease onset, age at death (if applicable), disease duration, sex, ethnicity (with any ancestry information), symptomatology, family history (including whether family carried the same variant or not), and any family consanguinity). Corresponding authors were contacted for reports with missing information, and all data was retrieved while maintaining patient consent and confidentiality. For consistency, we maintained each report's prediction of variant pathogenicity (even if it was contradicted in other studies) and included the predictions of gnomAD and ClinVar pathogenicity data to compare.
For meta-analysis, all normality and variance tests were performed in RStudio. To assess for normality, the Shapiro-Wilk test was chosen as it is more sensitive to deviations from normality and suitable for smaller sample sizes (our overall cohort was large, however, some sub-groups assessed were small (n < 30)). Though the main pool of patient data was the same, each group was assessed for normality for each pairwise comparison as the patient groups varied in size. If p ≤ 0.05 for the Shapiro-Wilk test (rejecting the null hypothesis indicating a non-normal distribution), the Wilcoxon rank-sum test (for dependent groups) or Mann-Whitney U test (for independent groups) was chosen for comparison analysis. If p > 0.05 for the Shapiro-Wilk test (failing to reject the null hypothesis and indicating the data does not significantly deviate from a normal distribution), the groups were tested for equality of variance using Levene's test. If p ≤ 0.05 for Levene's test (rejecting the null hypothesis indicating a significant difference in variances between the groups), Welch's t-test was chosen for comparison analysis. If p > 0.05 for Levene's test (failing to reject the null hypothesis and indicating no significant difference in variance between the groups), Student's t-test (for independent groups) or paired t-test (for dependent groups) was chosen for comparison analysis. All comparisons were performed in RStudio and Microsoft Excel using a two-tailed distribution to investigate differences in either direction. The significance threshold for all pairwise comparisons used was set at p < 0.05.
Results and discussion
We found partial information for 743 individual PINK1 cases (see Supplemental Information 1). A total of 280 patients were found with the minimum requirement for analysis (potentially pathogenic variant and at least one of sex or age at onset data). Their data are summarized in Table 1 (refer to Supplemental Information 3 for individualized data). For analysis, compound heterozygous variants were grouped with homozygous variants as they are generally accepted to have the same effect by mutating both alleles. Individualized age at onset data was found for 235 cases and individualized sex data was found for 252 cases, however it must be noted that specific variants were often reported inconsistently, and in single cases or low numbers overall. As such, our analyses associating observed differences in specific variants are based on the available data and highlight the need for consistent PINK1 PD testing and reporting.
Summary of PINK1 variants and case data for meta-analysis.
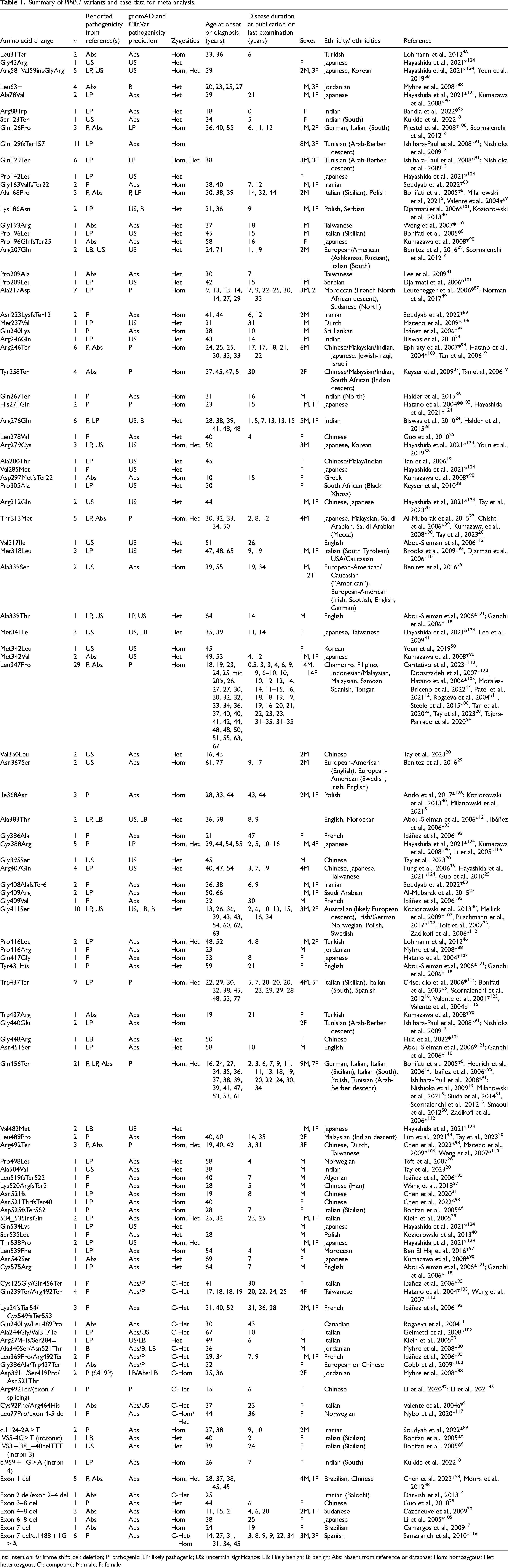
Ins: insertion; fs: frame shift; del: deletion; P: pathogenic; LP: likely pathogenic; US: uncertain significance; LB: likely benign; B: benign; Abs: absent from reference or database; Hom: homozygous; Het: heterozygous; C-: compound; M: male; F: female
Populations with the highest PINK1 variant rates are overlooked
From our systematic search, we found 279 cases with ethnicity information (Figure 1). Most reports of PINK1 variants were found in Asian and European patients (35.13% and 31.54%, respectively), followed by Africa (17.92%). PINK1 variants were most frequently reported in cases originating from Japan (12.19%), Italy (11.83%), and Tunisia (8.96%). However, when the case frequency was normalized to the population of the country of origin, 128 the countries with the highest frequency and incidence were Tonga (5.61E-05, 1:17,810), Guam (2.33E-05, 1:42,944) and Samoa (1.35E-05, 1:74,127) (Figure 1(b)).
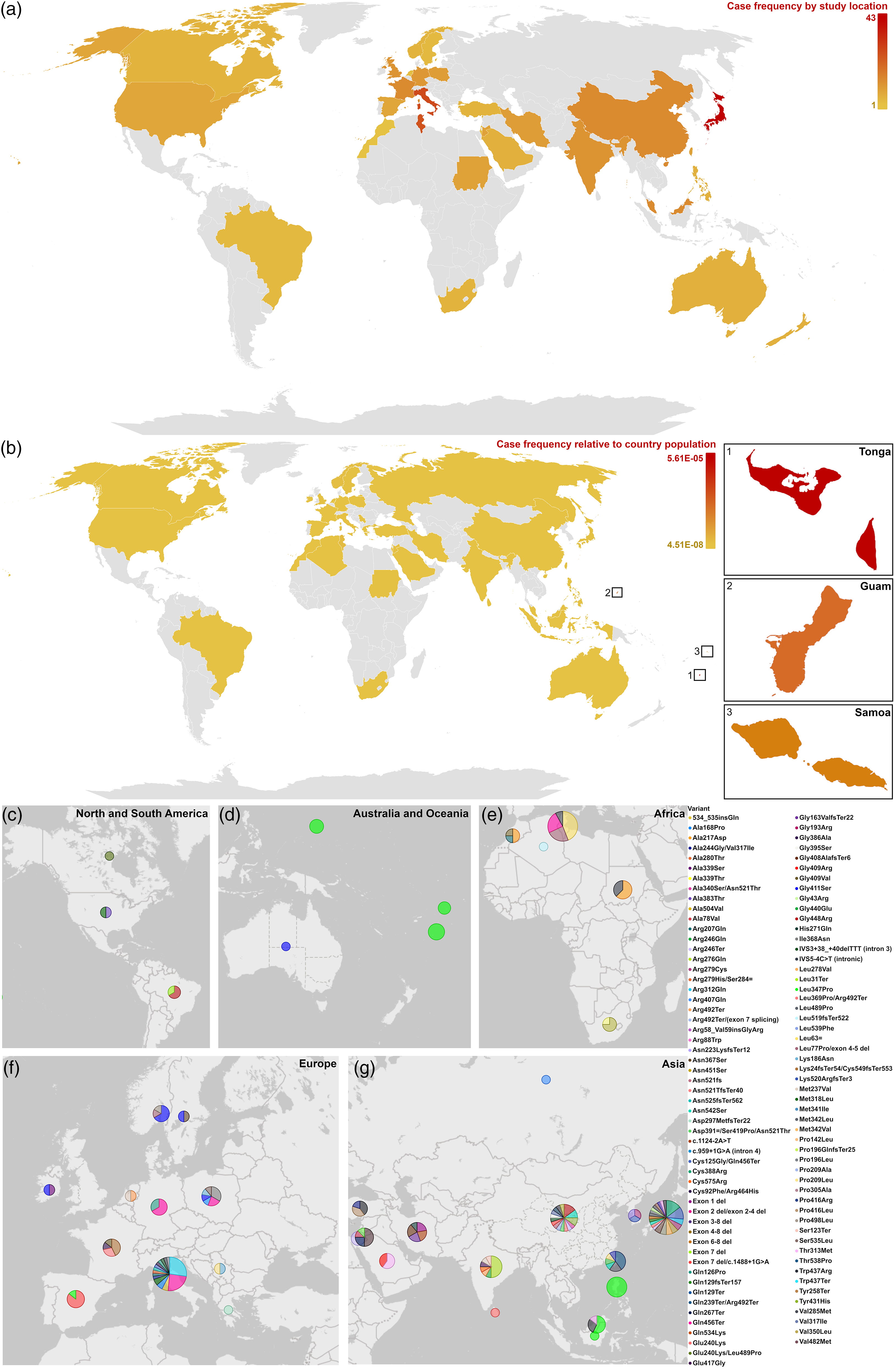
Case frequency of PINK1 variants across countries (n = 279). Overall case frequency by (a) study location, and (b) ethnic origin with frequency normalized to country populations (using World Bank Group data 128 ) with the top three countries enlarged. Case frequency by specific variant for (c) North and South America, (d) Australia and Oceania, (e) Africa, (f) Europe, and (g) Asia. Continents are not to scale between each map, but circles are to scale and represent n. Circles are in the country center and do not represent any specific region. Maps are configured to show only the countries with reported cases corresponding to the continent(s).
Specifically, the most frequently reported variants were p.Leu347Pro (pathogenic, n = 29), p.Gln456Ter (pathogenic, n = 21), and p.Gln129fsTer157 (uncertain significance, n = 11) (Figure 1(c)–(g)). Of the p.Gln456Ter cases, 71.43% were of European descent (Italian, German, Polish, English), with the remaining cases being of African descent (Tunisian). Of the p.Leu347Pro cases, 51.72% were of Asian descent (Filipino, Malaysian, Indonesian), another 44.83% were of Polynesian descent (Samoan, Tongan, Chamorro), and the remaining cases were European (Spanish). The worldwide allele frequency of p.Leu347Pro is 0.0016%, 129 but the frequency across reported Polynesian, Pacific and Southeast Asian populations is as high as 0.33–5%.11,12,86 Allele frequency here refers to the prevalence of an allele across all chromosomes in a population. The frequency of p.Leu347Pro for West Polynesia (Samoa, Tonga, Tokelau, and Niue) found in New Zealand by Patel et al. (2021) was 2.8%. Therefore, homozygous p.Leu347Pro variants may be implicated in approximately 1 in 1300 West Polynesians, increasing beyond the general definition of a rare disorder (approximately ≤1:2200).12,130 One possible explanation for these high concentrations of PINK1 variants is their spread through specific migration routes and genetic bottlenecking. 12 Archaeological evidence suggests ancient Guam DNA likely descended from Filipino ancestry, and this DNA is closely linked to early Lapita individuals from Vanuatu and Tonga. 131 The p.Leu347Pro variant thus could have been carried throughout Polynesia via a common founder population from Southeast Asia. 131 In the case of p.Gln456Ter, no established theories link the Italian, German, Polish, English and Tunisian populations. Still, a migratory explanation could be plausible due to the proximity of the countries (Figure 1(e) and (f)).
Altered patterns of PINK1 inheritance
In genetic bottlenecking, there is often a founder effect where the reproductively isolated population is subject to decreases in genetic diversity and increases in consanguinity, the coupling of people who share a common ancestor. 132 Consanguinity of people carrying PINK1 heterozygous variants increases the likelihood of offspring receiving both mutated copies and thus PD, as PINK1 follows an autosomal recessive inheritance pattern.2,132 The highest rate of PINK1 variants (8.9%) was found in a multicontinental cohort of Parkin- and DJ-1-negative consanguineous families with autosomal recessive PD. 105 We identified several consanguineous families from Sudan, Jordan, and Iran with the same inherited PINK1 variants.87–89 Homozygous variants p.Gln456Ter, p.Gln129Ter, p.Gln129fsTer157, and p.Gly440Glu (all pathogenic except p.Gln129fsTer157, a variant of uncertain significance) were found in 42 PD patients from 90 Tunisian consanguineous families. 13 A consanguineous German family was found with a high carrier rate of the p.Gln456Ter variant (4 homozygous cases and 6 heterozygous possible cases from 20 family members), 15 while two consanguineous Iranian families carried homozygous deletions of exon 4 or exon 5 (3 cases of each from 130 familial PD patients). 14 Furthermore, two consanguineous families with a pathogenic Trp437Ter variant in PINK1 share a common haplotype, implying common ancestry and a potential founder effect.114,115 In Italian patients with potentially pathogenic 534_535insGln variants, haplotype analysis indicated a plausible shared allele and a common founder. 39 In addition to published data, through additional requests for information we acquired consanguinity information for Polish p.Ile368Asn siblings (Zbigniew and Milanowski, 2024, personal communication), indicating consanguinity effects on PINK1-PD inheritance may be more common than reported.
The phenomenon of ‘pseudo-dominance’ has been observed when there is a high carrier rate of autosomal recessive disease variants, in which mating of an affected homozygote with an unaffected heterozygote mimics an autosomal dominant pattern (50% chance of offspring inheriting two mutated copies and thus PD, meaning ≥1 affected in 2 consecutive generations).132,133 Affected heterozygote patients with consanguineous parents in Italy and the Philippines have been described as exhibiting inheritance patterns suggestive of autosomal dominant transmission.6,113 The observed increase in ‘rare’ PINK1-positive Parkinson's disease (PD) cases within smaller consanguineous populations might be attributed to pseudo-dominance, as highlighted in the literature.6,16,90,113,114,122
Ethnic bias in PD research and diagnosis
This trend of geographical PINK1 ‘hotspots’ suggests a potential underrepresentation of specific populations in genetic research, particularly those outside European or industrialized nations, which may reflect a systemic research bias. For example, the West Polynesian ‘hotspot’ of pathogenic p.Leu347Pro was reported in Samoan and Tongan PD patients living in New Zealand and no research has been conducted in their countries of origin thus far. In our analysis, we observed that the countries that the studies were conducted in did not always correspond to the ethnicity of the cases, indicating a potential deficiency of access or resources in the locations of interest.
Despite recognizing clear familial patterns of PINK1 PD, the discovery of numerous variants unique to individual cases prompts further investigation into the significance of variant-specific differences and their locations on the PINK1 gene. The lack of standardized guidelines for genetic PD panel testing and a tendency to overlook ethnicity—though European labs tend to offer more comprehensive panels—underscores the need for more inclusive research and testing protocols.134,135 A recent large multi-center study advocating for better genetic testing reported low numbers of PINK1 PD but acknowledged that their cohort was predominantly European, 136 which shows that this lack of ethnic diversity is still a current issue requiring active and intentional attention. This can be achieved, however, as evidenced by an ongoing multi-center study working against this bias to characterize the global spectrum of monogenic PD. 137 Such measures are essential, especially for communities facing more significant healthcare challenges,138–141 to ensure a broader representation and understanding of PINK1 PD's genetic landscape.
Homozygous PINK1 variants are not all created equal: different effects on protein function influence overall PD risk
Function and structure of PINK1
The PINK1 gene encodes the mitochondrial serine/threonine-protein kinase PTEN-induced kinase 1, which regulates mitophagy and is protective in oxidative stress-induced apoptosis.142,143 Under normal circumstances, the PINK1 protein accumulates on the membranes of damaged mitochondria, recruiting and phosphorylating both ubiquitin and the Parkin protein to tag dysfunctional mitochondria for degradation. 144 A detailed discussion of PINK1's function in disease pathogenesis is available in Truban et al. (2017). 143 PINK1 has over 300 documented variants, from point variants to large gene rearrangements that can destabilize, truncate and inactivate the protein.144,145 Not all variants are pathogenic, so it is essential to classify individual variants and their consequences to inform disease prognosis. 145
The PINK1 protein consists of 581 amino acids, with a kinase domain (located between amino acids 156–511), a region for outer membrane localization (amino acids 111–117), and a disordered region (amino acids 189–208).146,147 Less well-defined regions include the early mitochondrial targeting sequence (between amino acids 1–80), subdivisions of the kinase domain called the N-lobe and C-lobe (between 156–340 and 320–511, respectively), and a C-terminal domain (between amino acids 486–581), though these are not established in protein databases and the literature varies.145,148–150
Mechanisms of PINK1 pathogenesis
Loss of PINK1's kinase activity likely initiates PD pathogenesis, though the significance of variants outside this kinase domain remains unclear.142,151,152 In vitro cell experiments demonstrate many mechanisms by which PINK1 variants may disrupt normal function depending on their location. Variants p.Glu240Lys (uncertain significance), p.Gly309Asp (uncertain significance) and p.Leu489Pro (likely pathogenic) spread wide across the kinase domain appear to destabilize the PINK1 protein and disrupt kinase activity in transfected human or animal cells, with p.Gly309Asp specifically disrupting adenosine diphosphate binding required for normal mitochondrial regulation.115,153 Cells from patients with p.Val170Gly variants (uncertain significance, absent from the ClinVar database), located relatively early along the PINK1 kinase region, show swollen mitochondria and dysregulated mitochondria function via the altered activity of respiratory chain enzyme complexes. 154 Patient cells with variants near the end of the kinase domain also show varying effects; p.Trp437Ter (pathogenic) downregulates expression of the S100A4 calcium-binding protein in mitochondria, leading to neuronal cell death, 155 while p.Gln456Ter (pathogenic) patient cells show decreased ATP levels, resulting in impaired bioenergetic function. 154 Overall, there is not one common mechanism through which pathogenic PINK1 variants initiate PD-related pathogenesis. Instead, a varied spectrum of alterations to mitochondria and cell activity may disrupt PINK1 function and cause or contribute to disease. 122
A spectrum of PINK1 pathogenicity
One systematic analysis assessed 50 missense PINK1 variants and re-classified their pathogenicity using published clinical, population-wide and functional evidence, combined with additional functional experiments. 145 In standard terminology, variants are categorized depending on the available evidence ranging from ‘benign’ or ‘likely benign’ to ‘likely pathogenic’ or ‘pathogenic’ or as a ‘variant of uncertain significance’, considering the strength of their segregation with the disease, minor allele frequency, and homozygosity in controls. 145 From the systematic analysis, most pathogenic variants were located in the kinase region, 145 paralleling our present data showing approximately 90% of pathogenic or likely pathogenic variants lay within this domain (Figure 2), reiterating the importance of the kinase in normal PINK1 function. The specific variant had a clear effect on mitophagy; most variants classified as pathogenic or likely pathogenic caused a partial or total reduction of normal PINK1-related mitophagy, indicating this loss of function is a primary mechanism of PINK1-related PD. 145
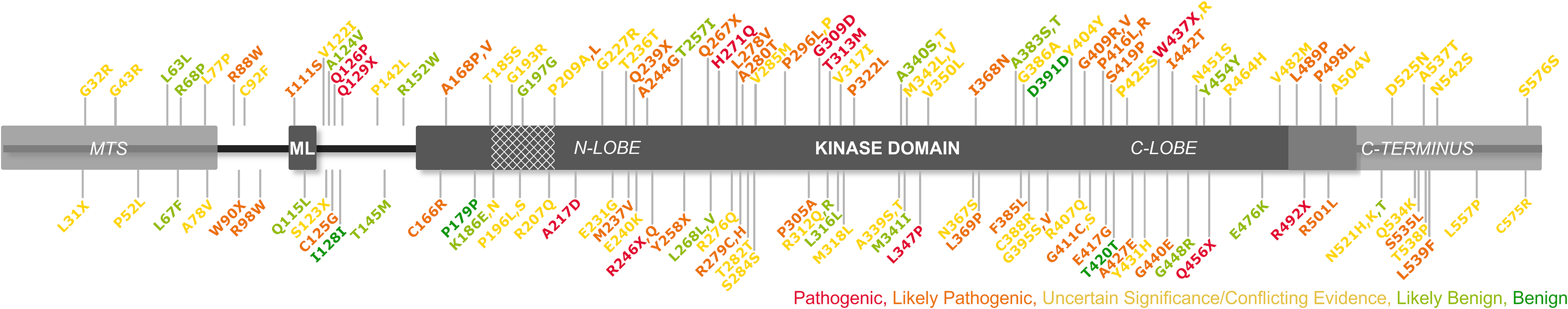
Gene location of PINK1 point variants. Pathogenicity is described using available information from the original studies and the gnomAD/ClinVar database. Variants described using amino acid single letter codes (International Genetic System). Cross-hatched area: disordered region; italicized labels: regions not well-established in databases or literature; ML: region for outer membrane localization; MTS: mitochondrial targeting sequence.
Only some of the variants found in our systematic search were included in the large-scale analysis by Ma et al. (2021). 145 Still, those assessed were all predicted pathogenic, likely pathogenic, or of uncertain significance. For variants predicted as being of uncertain significance, the functional assays indicate that though they do not entirely abolish kinase activity and mitophagy induction, these variants may still have a partial impact. For example, p.Pro296Leu and p.Gly309Asp, located in the gene region involved in ubiquitin binding, caused some disruption to mitophagy via the ubiquitin-PINK1-Parkin interaction and thus linked their position directly to their function. 145 Other variants reported as benign (p.Gln115Leu, p.Ala340Thr, and p.Asn521Thr) also reduced mitophagy induction, though not to a pathogenic level, suggesting a certain level of dysfunction is tolerated until a threshold at which pathogenesis is initiated.145,156
Potential heterozygous PINK1 insufficiency
Our observation of PINK1 PD cases with non-benign heterozygous variants,9,11,17,79 including when other risk genes (Parkin, DJ-1) are eliminated as a cause,6,18,19 suggests that even one healthy allele may not meet the threshold for normal function. Haploinsufficiency is when the 50% level of function produced by the healthy gene copy is inadequate to produce the normal phenotype, which is classically a trait of autosomal dominant genes but can manifest in autosomal recessive genes. 156 In cell lines expressing one mutant copy of PINK1, there is decreased dopaminergic cell survival compared to the wild-type, and cell lines expressing two mutant copies have even lower survival. 157 Five PINK1 heterozygous variants (p.Tyr431His, p.Asn451Ser, p.Glu476Lys, p.Arg501Pro, and p.Cys575Arg, all variants of uncertain significance absent from the ClinVar database except the likely benign p.Glu476Lys) showed decreased mitochondrial membrane potential in vitro compared to wild-type PINK1. 121 Therefore, variants in PINK1 may act as risk factors and increase the likelihood of more severe disease, with one mutant copy causing an ‘intermediate’ phenotype. Insufficient or excessive protein levels can alter the gene dosage stability, leading to detrimental effects.158–160 Stochasticity in whether one healthy allele's function is above the threshold for initiating normal processes can differ from cell to cell, further complicating this balance.159,161–163 Heterozygous p.Gly411Ser variants (uncertain significance) in PINK1 decrease kinase activity in vitro compared to controls, and p.Gln456Ter (pathogenic) heterozygotes induce an inhibiting effect, not simply a loss-of-function. 122 Thus, it is apparent that PINK1 variants and their zygosity differentially affect the normal protein function and contribute to an individual's overall PD risk in a variant-specific manner.
Heterozygous PINK1 brains show significant but subclinical signs of disease
One of the most repeated arguments for heterozygous PINK1 variant risk in PD is the observation of detectable differences in neuroimaging data compared to healthy controls. Structural abnormalities in PINK1 heterozygotes include increased substantia nigra echogenicity, which is indicative of higher iron levels and posited as a vulnerability marker for PD.164,165 In animal studies, heterozygote PINK1 knockout mice were unable to produce long-term potentiation despite repetitive cortical inputs to striatal neurons, likely related to an observed decrease in striatal dopamine release. 166 In human patient positron emission tomography, PINK1 homozygotes exhibit a 60–85% decrease in 18-fluorodopa uptake in the caudate and putamen in comparison to controls, while heterozygotes exhibit a 20–30% decrease, indicating a subclinical but significant reduction of striatal dopamine storage capacity.167,168 Another imaging study observed increased grey matter volume in the putamen and internal globus pallidus in those with heterozygous PINK1 variants compared to controls, suggesting this chronic dysfunction was a compensatory mechanism for dopaminergic denervation. 169 This ‘compensation’ hypothesis was corroborated with functional data by the same research group showing abnormal recruitment of the rostral supplementary motor areas and rostral dorsal premotor cortex in PINK1 heterozygotes when they completed a simple motor task. 170 These studies present an argument consistent with the idea of haploinsufficiency and a threshold for disease, in which heterozygous PINK1 can cause significant brain changes even if they are not detected in routine clinical observation. However, the same few studies are repetitively cited, and their arguments should be strengthened through newer studies in large cohorts.
Heterozygous PINK1 variants do not cause PD (alone)
Many support the dogma that PINK1 PD has autosomal recessive inheritance, arguing that there is little to no evidence for a causative role of heterozygous PINK1 variants.20,21,91,92,119,171,172 Meta-analyses in large PD case-control datasets detected no apparent association between heterozygous variants and PD in European119,171 and North American populations. 93 Overall, heterozygous PINK1 variants appear to have a low (1–25%) penetrance 156 and might play only a minor susceptibility role in PD pathogenesis. 92
Our systematic search found 378 unaffected carriers reported with PINK1 variants (see Supplemental Information 4). Of these, 271 were classified as non-benign, and 90.04% carried heterozygous variants. Only 38 individuals carried variants predicted as pathogenic or likely pathogenic in EOPD by both the original report and gnomAD/ClinVar, and all were in heterozygous forms (p.Gln129Ter, p.Ala217Asp, p.Arg246Ter, p.Thr313Met, p.Leu347Pro, p.Trp437Ter, p.Gln456Ter, and p.Arg492Ter). Sixteen of these patients had sex recorded (6 males, 10 females), while 10 had a recorded age at observation that ranged from 39–86 years (mean ± SEM = 68.30 ± 4.65). Compared to affected patients, 15 cases carried these same variants in heterozygous forms (p.Thr313Met, p.Gln129Ter, p.Leu347Pro, p.Trp437Ter, p.Gln456Ter, and p.Arg492Ter). From 8 affected cases with age at onset data, the span was 34–77 years (mean ± SEM = 47.75 ± 6.05).
As the mean age of the unaffected carriers is over 50 years and higher than that of affected patients at onset, it could be assumed that these heterozygous variants do not pose a significant risk factor for developing EOPD in these individuals. Yet, affected and unaffected heterozygotes can present with identical PINK1 variants, even within the same family.16,41,114 This questions whether other factors, aside from variant-specific differences and random chance, influence whether PINK1-related PD manifests.
Other variants of mitochondria function influence PINK1 disease
Research from the fruit fly Drosophila melanogaster has identified other mechanisms that may determine whether heterozygous PINK1 variants are significant enough to manifest disease by exacerbating deficiencies or compensating for them. A typical Drosophila melanogaster PINK1 mutant (PINK1B9 is commonly used) shows muscle degeneration in the form of impaired flight and usually a mitochondrial deficiency in ATP production.173–175 Alterations to mitochondria-related elements like transport, degradation, morphology, integrity, bioenergetic function, and antioxidant pathways can rescue the observed Drosophila melanogaster deficits via manipulation of kinases, proteases, signaling receptors, scaffolding proteins, vitamins, and detoxifying enzymes.173,176–186 Changes to gene expression or generation of these products from other variants or cellular signals could modulate the effect of a PINK1 variant, even if only slightly; it may be enough to tip the scale. Though performed in Drosophila melanogaster, these experiments provide plausible modifiers of PINK1 disease that may contribute to the varying presentations observed in heterozygous carriers.
Environmental toxins are risk factors in combination with PINK1 variants
Environmental toxins like paraquat, rotenone, and manganese (found in insecticides, herbicides, and working in mining and steel mills) can cause oxidative stress, acceleration of motor deficits, and increased dopaminergic neuron desensitization and death in PINK1 PD in vitro and in vivo.187–192 Five early-onset PINK1 heterozygotes in a population of Indian PD patients (with variants p.Arg246Gln (likely pathogenic) and p.Arg276Gln (uncertain significance)) were all described with additional marked and rare symptoms of lower limb pain, speech impairment, and impaired ocular movement. 24 Notably, these patients had overall poor responses to levodopa, which is in contrast to most PINK1 patients, and they were also exposed to environmental toxins for up to 25 years (pesticides, insecticides, or living in rural areas drinking water from a well). 24 Brazilian patients with a PINK1 single nucleotide polymorphism (IVS1−7A > G) who were exposed to environmental risk factors (rural living, drinking water from wells, or exposure to pesticides, insecticides, or organic solvents) had a lower age at disease onset compared to other EOPD patients, possibly via alterations to gene expression (as the polymorphism is located in the intronic region of PINK1). 82 Different PINK1 variants exhibit different molecular effects, which can subsequently be exacerbated by environmental toxins, resulting in a significant risk factor.
A common but distinct PINK1 phenotype
Though the reports in our analysis varied greatly from detailed case studies to simple clinical notes, the 193 patients with symptom information presented similarly to past descriptions of PINK1-PD, exhibiting frequent occurrences of gait impairment (33.16%), often severe, indicating symptoms predominated in the lower limbs6,8,9 (see Supplemental Information 5). Asymmetry of symptoms was observed in almost half of the patients (44.04%), and occurrences of early dystonia (12.44%) and psychiatric symptoms (up to 18.14%) were consistent with the literature.1,2,7 PD patients with PINK1 variants may be more likely to develop psychiatric disturbances compared to those with idiopathic PD.6,94,95,105,114,193 As expected from previous reports,6,7,9 most patients responded to L-dopa (74.09%) to a very good or excellent degree (23.78% of responders) with doses often around 300–600 mg daily, though predictably, many also presented with L-dopa-induced dyskinesia and motor complications (35.75% and 19.69%, respectively). It is suggested that PINK1 PD may have significantly higher rates of dyskinesia and motor fluctuations compared to other monogenic forms of EOPD. 194
Anosmia has been observed in a homozygous Filipino case of pathogenic p.Leu347Pro PINK1 PD, 120 four pathogenic p.Gln456Ter homozygotes (and seven heterozygotes) from a large German family, 167 and across several Italian families, in one likely pathogenic p.Ala168Pro homozygote and six pathogenic p.Trp437Ter homozygotes. 123 Of note was that eight unaffected carriers of heterozygous p.Ala168Pro or p.Trp437Ter from the same families also showed mild to moderate hyposmia at a pathologically significant level for their age, highlighting the importance of this symptom in early PD detection. 123
Sex and zygosity in the PINK1-PD symptom profile
Across the sex groups and zygosity groups, the mean years of disease duration appeared slightly longer for females compared to males (17.06 ± 1.32 years (n = 68) and 13.43 ± 1.13 years (n = 75), respectively) and homozygotes compared to heterozygotes (17.42 ± 1.13 years (n = 105) and 11.91 ± 1.05 (n = 57), respectively). Males, on average, appear more likely to receive a higher dose of L-dopa than females and may be more likely to receive more than one other course of treatment (see Supplemental Information 6). Additionally, they appear more likely to be reported with more severe depression or mood disturbance. Homozygotes may be more likely to present with gait impairment and dystonia compared to heterozygotes (see Supplemental Information 7). They may also receive a higher L-dopa dose than heterozygotes, which would be expected if homozygous disease is assumed to be more severe. These findings are novel, as previous PINK1 PD research either lacks the cohort size to perform any quantitative comparisons, or instead focuses broadly on the disease rather than any patient-specific differences.
However, it must be acknowledged that heterogeneity of reporting (likely omitting many symptoms) and a broad range in disease duration (0–47 years, likely changing over time) limit the definitive conclusions that can be drawn about symptomatology. Overall, this calls for more systematic reporting of patient symptom information and a shift in the perspective of PINK1 PD from a homogenous rare disorder to a multi-faceted spectrum of disease.
Age at onset: an intermediate phenotype for heterozygous PINK1?
PD-affected PINK1 heterozygotes are frequently described with higher ages of onset than PINK1 homozygotes and a phenotype akin to idiopathic PD.6,25,26,90–92,118 According to published literature, the mean age at onset for patients with homozygous PINK1 variants is 33 years, 195 compared to approximately 60–65 years for idiopathic PD. 196 Our systematic search found age at onset information for 216 patients with potentially pathogenic PINK1 variants (38.41 ± 0.91, mean ± SEM). The average age at onset for homozygous and compound heterozygous variants (n = 140) was 35.96 ± 1.12 years (mean ± SEM), while for heterozygous variants (n = 74) it was 43.36 ± 1.58 years (mean ± SEM) (Figure 3(c)). The mean age at onset for heterozygotes is still early-onset (<50 years of age), yet it is significantly older than the mean for homozygotes (Student's t-test, p = 0.0000930). This is consistent with the theory of an intermediary level of heterozygous disease between severe early-onset homozygotes and late-onset idiopathic patients, manifesting as a mixed PINK1-idiopathic phenotype.
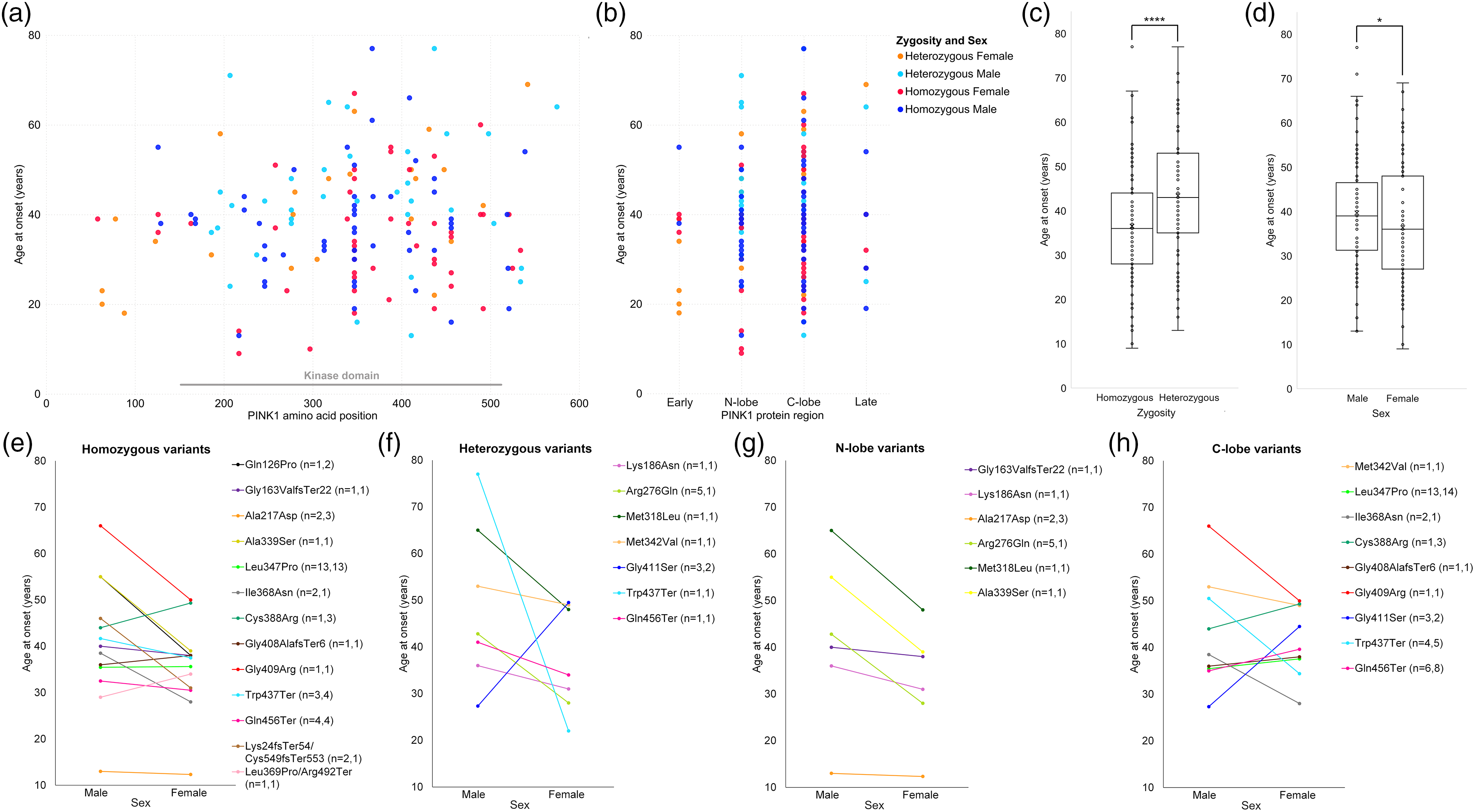
Meta-analysis of 192 PINK1 cases detailed in Table 1 stratified by zygosity, age of onset and sex. Cases with single point variants, by (a) amino acid position, and (b) gene region. Age at onset by (c) zygosity and, (d) sex. Age at onset by sex of variants reported in both sexes, for (e) homozygous variants, (f) heterozygous variants, (g) variants in the N-lobe of PINK1's kinase domain, (h) variants in the C-lobe of PINK1's kinase domain, n = males, females.
Females have earlier ages at onset than males
The overall mean age at onset for males (n = 99) was 39.71 ± 1.34 years and 35.5 ± 1.42 years for females (n = 90), with a statistically significant difference between sexes (Student's t-test, p = 0.0316) (Figure 3(d)).
To establish if PINK1 variant location is associated with age of onset and sex, we categorized the variants into protein regions in Figure 3(b) (excluding compound variants as they do not have a single location). We defined these as ‘early protein region’ (amino acids 1–155, comprised of the mitochondrial targeting sequence and other regulatory regions), N-lobe of the kinase domain (156–340), C-lobe of the kinase domain (341–511), and ‘late protein region’ (512–581) based on the varied lengths described in the literature.145,146,148–150 Of these variants, 86.98% lay within the kinase region (Figure 3(a) and (b)), reemphasizing the association of variants in this domain with disease.
Sex differences are dependent on zygosity and variant location
To further tease apart trends in sex differences, we averaged the ages at onset for the specific variants reported in both sexes (Figure 3(e)–(h)). Homozygous variants reported in both sexes showed females have significantly earlier ages at onset than males (paired t-test, p = 0.0362). Variants in the N-lobe of the PINK1 kinase domain also showed this pattern of significantly earlier onset ages for females compared to males (paired t-test, p = 0.0293). Six out of seven heterozygous variants showed a trend for lower ages of onset in females than males, though pairwise comparison was not significant (paired t-test, p = 0.234). Variants in the C-lobe show more variation and not as clear of a sex difference (paired t-test, p = 0.654).
Sex effects are different for PINK1-PD versus idiopathic PD
Sex effects have been established for general PD risk in previous literature, with the incidence of PD in males being 91% higher than females, most prominent after 60 years of age. 197 Males, in general, also have earlier onset ages than females. 198 However, little to no literature exists about the link between sex and PINK1 PD specifically. In PINK1-knockout rodents, downregulation of neurogenesis 199 and several hundred other genes occurred in both sexes, though there was some variance in gene type (genes involved in axon guidance, syntaxin, and ubiquitin were predominantly affected in male rats versus genes involved in sodium channel activity, calcium signaling, and synapse organization in females). 200 In aged mice, PINK1 is upregulated with no apparent sex differences, 201 but in humans, PINK1 upregulation in the substantia nigra of PD patients is only observed in males. 202
Though overall animal models suggest sex differences on a genomic level, there are always constraints to translating these data to humans. In our systematic search, we observed a similar incidence of males and females with PINK1 variants (140 males and 124 females over the whole cohort), though we identified a clear pattern of earlier age at onset for females compared to males, with further analyses suggesting zygosity of the variant and the location along the gene may also play a role. This finding is novel and of particular interest as the relationship is the inverse to that seen in idiopathic disease. Continued work to document additional PINK1 PD cases will enable the validation of this finding and the investigation of any other differences in clinical profiles.
α-Synuclein pathology is evident in PINK1 disease
The pathology of PINK1 PD is under debate, and due to a lack of extensive research, it is assumed that α-Syn is not implicated in the disease. However, this may be an incorrect assumption. Typical idiopathic PD pathology is characterized by misfolded and aggregated α-Syn proteins, which form toxic Lewy bodies that slowly spread throughout the brain, 203 and changes in PINK1 may be related to α-Syn accumulation. α-Syn aggregates are present in PINK1-deficient rodents, 204 and this absence of functional PINK1 increases vulnerability to α-Syn aggregation compared to control rodents.205,206 The PINK1 protein is hypothesized to promote the degradation of excess α-Syn, 207 and different variants in PINK1 may produce varying pathology. Amino acid deletion at position 245 of the protein (pathogenic), variant p.Leu347Pro (pathogenic), and variant p.Glu417Gly (likely pathogenic) in PINK1 cell cultures caused an increase in α-Syn aggregation, 208 and notably, p.Gly309Asp (uncertain significance) completely abolished PINK1's interaction with α-Syn to initiate autophagy. 207 Fibroblasts from patients with PINK1 p.Gly309Asp variants had increased α-Syn expression compared to controls, and this effect was replicated in control fibroblasts by knocking out PINK1, highlighting the importance of PINK1 in α-Syn degradation. 209
α-Synuclein pathology in homozygous evidence
Though limited, the human post-mortem pathology of homozygous PINK1-affected brains points towards α-Syn involvement (Table 2). In a Norwegian case with over 30 years of disease duration, Lewy pathology was observed in the temporal cortex, locus coeruleus, parahippocampal cortex, and substantia nigra, 117 while a younger Spanish case with eight years of disease duration before death at 39 years of age was positive for Lewy bodies in the reticular nuclei, Meynert nucleus, and substantia nigra. 116 A case from Guam was diagnosed with PD at a surprisingly late age of 67 years, and postmortem observations found Lewy pathology only in the amygdala, in contrast to the previous reports where this region was spared. 86 However, this Guamanian patient was diagnosed with amyotrophic lateral sclerosis at 44 and also carried a heterozygous pathogenic DNAJC13 variant, so their results must be interpreted with caution. 86 The only record arguing for the absence of Lewy bodies in PINK1 PD describes a Japanese case with 10 years of disease duration and death at 64 years of age. 127 All three of these α-Syn-positive cases described had substantia nigra neuron loss and gliosis consistent with typical PD pathology.86,116,117
Postmortem analyses of α-synuclein in PINK1-mutated Parkinson's disease cases.
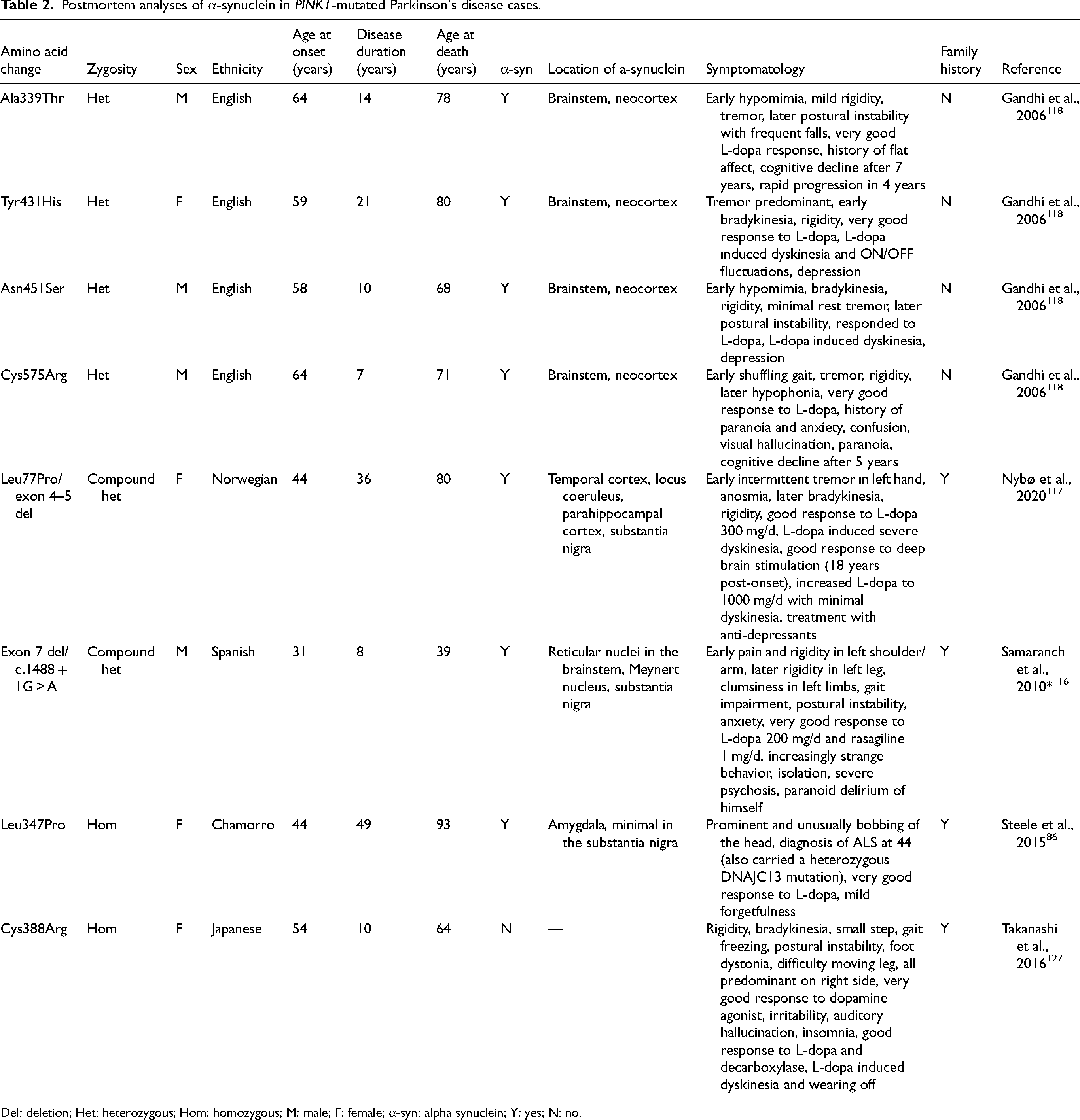
Del: deletion; Het: heterozygous; Hom: homozygous; M: male; F: female; α-syn: alpha synuclein; Y: yes; N: no.
α-Synuclein pathology in heterozygous evidence
Four heterozygous PINK1 cases have been investigated post-mortem for Lewy bodies, and α-Syn pathology was present in the brainstem and cortex 118 (Table 2). Though Lewy body pathology cannot be confirmed until after death, the diagnostic PD test 123 meta-iodobenzylguanidine (MIBG) myocardial scintigraphy assessing heart function is used as a proxy tool. 210 Decreased MIBG uptake reflects increased cardiac denervation, linked to subsequent sympathetic ganglia cell death and Lewy pathology. 211 PINK1 heterozygotes may have a decreased MIBG uptake compared to PINK1 homozygotes, similar to levels seen in idiopathic PD patients.90,124
α-Synuclein pathology in PINK1
Whether PINK1 PD causes α-Syn pathology to manifest or if pathology is a common effect regardless of PINK1 variants must be questioned. The pathology in the four heterozygote postmortem analyses was reported at similar levels to idiopathic PD cases. 118 Thus, it is unclear whether PINK1-related pathology involves Lewy bodies or if PINK1 heterozygous variants are coincidental and the pathology reflects a typical idiopathic phenotype. One α-Syn-positive homozygous case (Table 2) had a short disease duration (8 years) and died at the age of 39, which suggests the observed pathology was not simply a result of expected age-related degeneration. An interconnected relationship between α-Syn proliferation and PINK1-dependent mitochondrial function has been observed via TNF-receptor-associated protein 1, which interacts with PINK1 in mitochondrial regulation and mitigating α-Syn toxicity. 185 As well as debating the role of α-Syn in PINK1 PD, the reverse has been investigated, i.e., the role of PINK1 in the formation of α-Syn. In post-mortem brain tissue from confirmed α-Syn-positive patients, the mutated PINK1 protein was found to be a component of glial cytoplasmic inclusions (in MSA) and Lewy pathology (in PD and dementia with Lewy bodies). 212
Overall, α-Syn pathology was observed across ages and several different PINK1 variants, suggesting aggregation is not a specific effect of a given variant. Based on the limited data, 87.5% of PINK1 PD showed a presence of α-Syn aggregation, implicating α-Syn in PINK1 PD and disease pathology.
Conclusion
PINK1 PD manifests with a distinct phenotype of lower limb symptoms, a long-term excellent response to L-dopa, and a markedly early age at onset in contrast to idiopathic PD. However, PINK1 shows evidence for the presence of α-Syn pathology in disease progression, which idiopathic PD typically also exhibits. Despite similar observed presentation of PINK1-mutated PD regardless of variant zygosity and patient sex, disease onset is earlier for homozygotes versus heterozygotes and females versus males, indicating a spectrum of phenotypes within PINK1 disease. This spectrum has yet to be fully explored, especially in geographic regions underrepresented in research such as Polynesia, where we observed the highest variant rates.
As PINK1 PD is early-onset and progresses slowly, the appropriate disease management should differ from those diagnosed with late-onset idiopathic disease. Yet, the approach to all PD, including PINK1 PD, is often the same, and genetic testing is not always commonplace. In continued testing, treating PINK1 disease as a ‘sub-disease’ within PD will help establish and develop distinct phenotypic profiles of each specific variant. This approach is crucial for improving care for these individuals who face long-term disease burdens, especially in disadvantaged places where healthcare research and access are most vital.
Supplemental Material
sj-docx-1-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-docx-1-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-2-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-2-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-3-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-3-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-4-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-4-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-5-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-5-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-6-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-6-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Supplemental Material
sj-xlsx-7-pkn-10.1177_1877718X241304814 - Supplemental material for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology
Supplemental material, sj-xlsx-7-pkn-10.1177_1877718X241304814 for Rethinking ‘rare’ PINK1 Parkinson's disease: A meta-analysis of geographical prevalence, phenotypic diversity, and α-synuclein pathology by Eden Paige Yin and Birger Victor Dieriks in Journal of Parkinson's Disease
Footnotes
Acknowledgments
The authors acknowledge authors of studies included in the meta-analysis for their generous contributions of additional data: Dr Łukasz Milanowski, Dr Monika Rudzińska, Dr Joanna Suida, Dr Bruno Benitez, and Professor Christine Klein.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: EPY is funded through a University of Auckland Doctoral Scholarship. BVD is funded by a Health Research Council Hercus Fellowship (21/034), the School of Medical Science, the University of Auckland, and Te Tītoki Mataora (3729090 & 3729858). The funders had no role in study design, data collection and analysis, publication decisions, or manuscript preparation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available within the article and/or its supplemental material.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.