
Abstract
In 2023, a workshop was organized by the UK charity Cure Parkinson's with The Michael J Fox Foundation for Parkinson's Research and Parkinson's UK to review the field of growth factors (GFs) for Parkinson's disease (PD). This was a follow up to a previous meeting held in 2019. 1 This 2023 workshop reviewed new relevant data that has emerged in the intervening 4 years around the development of new GFs and better models for studying them including the merit of combining treatments as well as therapies that can be modulated. We also discussed new insights into GF delivery and trial design that have emerged from the analyses of completed GDNF trials, including the patient voice, as well as the recently completed CDNF trial. 2 We then concluded with our recommendations on how GF studies in PD should develop going forward.
Plain language summary
For many years, scientists have explored the idea of administering growth factors to the brain, thereby repairing the damage associated with neurodegenerative diseases like Parkinson's disease (PD). Growth factors like glial cell line-derived neurotrophic factor (GDNF) have shown promise in animal models of PD, and initial clinical trials suggested that this approach may be beneficial for patients. However, there has been some uncertainty in the field after a number of clinical trials did not reach their primary endpoints. Nevertheless, the development of growth factor therapies for PD has continued, with new trials underway. Recent developments in this scientific area were discussed at a workshop organized by a number of PD charities in 2023. The discussion and conclusions from that workshop are presented in this new paper.
Introduction
The rationale for using growth factors (GFs) for treating Parkinson's disease (PD) is that they can rescue and regenerate discrete neuronal populations which in the case of PD is most notably the dopaminergic nigrostriatal pathway. This has been trialed over the years using a number of agents of which the most frequent has been glial cell line-derived neurotrophic factor (GDNF) and related neurturin (NRTN) owing to its potency on the cognate receptor, GFRa1. These trials have shown some evidence of biological engagement with the target cellular population in the PD brain but without showing consistent major clinical benefits, and the reasons for this were discussed in our previous workshop in 2019. In summary, the inconsistency of effect for these GFs appeared to relate to the type of therapeutic product given, disease stage treated, delivery method including the volume of distribution across the target structure (i.e., caudate-putamen) of the agent, effective bioavailable concentration as well as trial design and the nature of the primary outcome measures. 1
Over the last 4 years this area of therapeutic interest in PD has continued, including new trials with cerebral dopamine neurotrophic factor (CDNF) 2 as well as GDNF gene therapy 3 and developments around improved long term delivery of therapeutic agents to the brain. 4 Linked to this has been the patient voice and their role in contributing to the debate, especially those who were in receipt of GDNF therapies in the past. This formed an integral part of this workshop especially given that they are able to provide valuable contributions to trial design and information materials, the risk/benefit consideration and support requirements for sustained trial retention going forward 5 as well as getting any drug licensed. 6 Furthermore, many of the experiences of previous trial participants captured through studies such as LEARN (ListEning to pARticipants of Neurosurgical trials), led to expressions of positivity to further research and trials with GFs, despite the fact that previous studies did not meet their primary end points. It is also worth noting that while single voices relate powerful individual stories, systematic evidence collection across a group of participants produces a more robust and complete picture of their experiences with actionable insights. In this respect, in-depth interviews of participants in the Bristol GDNF infusion trial have highlighted specific areas of trial design that were problematic for participants (Drew et al., in process, and personal communication within the workshop from a trial participant/caregiver and co-authors to this paper). Despite very varied experiences, many described difficulties in undertaking the motor assessments and also brain imaging, often compounded by the need to be in the clinically defined ‘off’ state for the assessments (which also necessitates travelling to the clinic in ‘off’, all of which can have a profound impact on the patient and family members). ‘Off medication’ assessments have been identified by colleagues in both pharmacological and non-pharmacological interventional studies as being an area of concern for patients and caregivers at recruitment and a challenge throughout clinical studies.7,8 Participants also often described a range of emotional impacts of the trial, importantly the psychological hurdle of the trial finishing and no longer receiving the treatment, whilst still having the surgical implant.
In this new commentary, we have sought to systematically address a number of key issues linked to this and the translation of GF therapies to patients (see Table 1). We have then integrated the conclusions from this into a wider debate around the future of GF clinical trials.
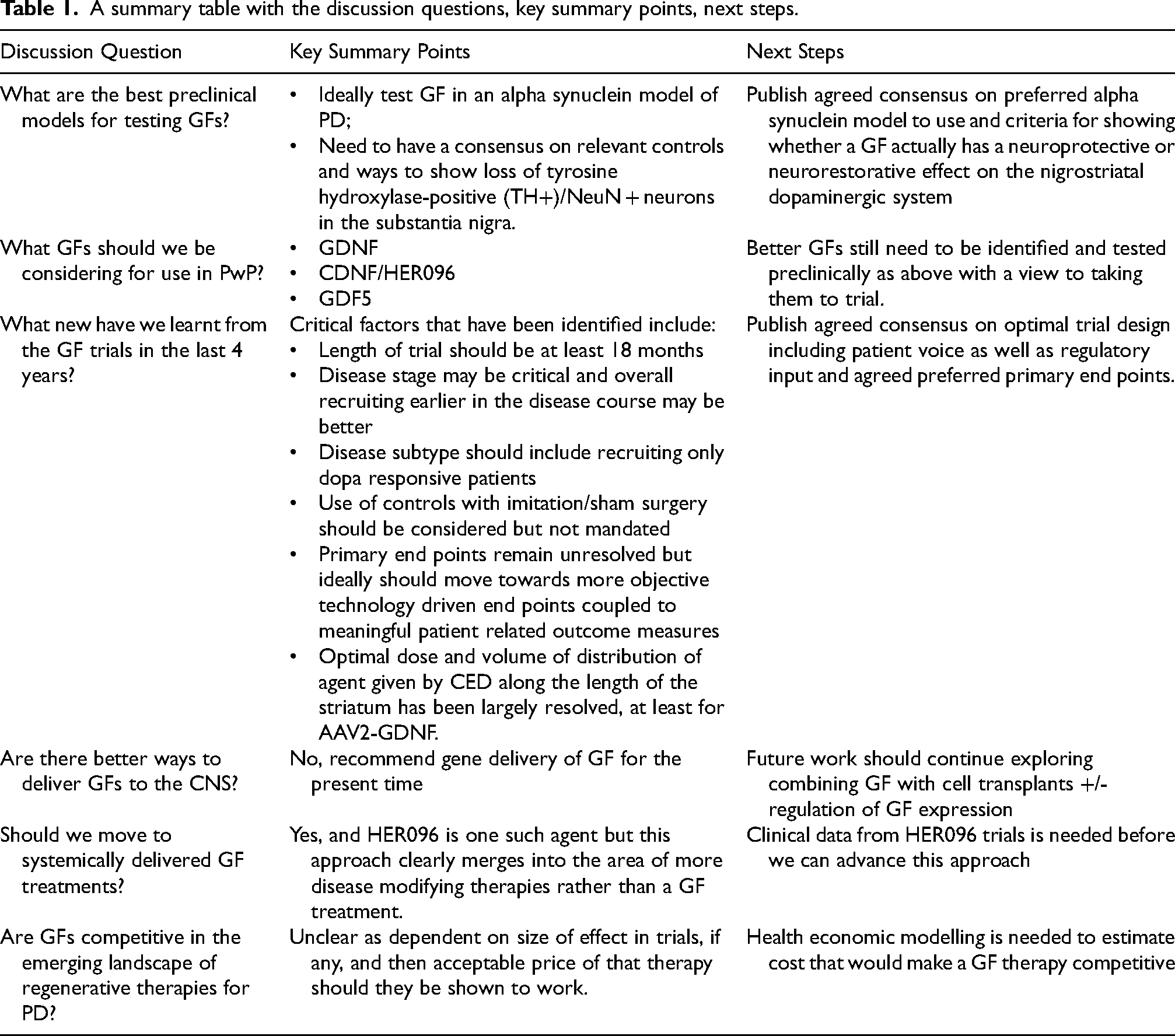
A summary table with the discussion questions, key summary points, next steps.
Preclinical models for testing GFs: where are we?
It is widely acknowledged that the well-established toxin-based models of PD have been effective at identifying and testing dopaminergic therapies, including cell based treatments. 9 But for putative disease-modifying strategies such as GFs, these are inadequate, since they do not consistently recapitulate many of the neuropathological features of PD, most notably pathogenic α-synuclein aggregation and chronic cellular dysfunction and loss across a number of neural networks. 9
To rectify this shortcoming, models using adeno-associated virus (AAV) vector-delivered α-synuclein were developed.10–14 However, one disadvantage of this model is the heterogeneity in behavioral deficits that are reported, probably resulting from differences in the viral serotype, titer, promoter, injection volume, levels of expression and other key methodological parameters.12,13 An alternative mouse model of α-synucleinopathy that has been developed relies on the injection of α-synuclein preformed fibrils (PFFs).15,16 This approach yields pathology that more closely reproduces that seen in the post mortem human PD brain tissue, including the appearance of α-synuclein inclusions similar to Lewy bodies. 17 As such, this model may be a better way by which to evaluate novel PD therapeutics, including GFs and in this respect a recent study adopting this model has reported showing a protective effect for intrastriatal CDNF motorically, although this did not reduce the number of phosphorylated α-synuclein inclusions found in the substantia nigra. 18 Using this same model, GDNF did prevent the accumulation of misfolded α-synuclein in DA neurons especially when the expression of Ret was shown to be constitutively active, 19 which was different to that seen when GDNF was tested in AAV α-synuclein models.12,13
Although this new PFF α-synuclein model is widely used, it is important to recognize that a wide range of PFF conformational species are used in these studies, sometimes in combination with AAV-α-synuclein—as such it is not surprising that many different outcomes have been reported depending on the exact protocol used.20,21 In addition, other combination models have been used including the addition of α-synuclein aggregators (FN075), 22 or α-synuclein modalities in conjunction with other dopaminergic toxins to replicate both the α-synucleinopathy and the neuronal deficit. 23 As the diversity of models used affects the rate of pathogenicity, it is challenging to compare the relative potency of neuroprotective GFs in these different models.
Much of this work, though, has provided helpful insights into GF function but one of the major issues has been that α-synuclein can downregulate the expression of phenotypic markers of dopaminergic neurons in the primate and human substantia nigra. 24 Thus, there is a need to establish appropriate controls, in order to allow for consistency and comparison between studies.
In summary, given all of this, it is timely for the field to develop a standardized set of reporting guidelines to be used in pre-clinical testing of GFs to distinguish between neurorestoration of phenotype versus true neuroprotection, while recognizing these may not be mutually exclusive. Core to this would be a consensus on the appropriate controls needed for each of these different models, as well as establishing consistency in the estimation of loss of tyrosine hydroxylase-positive (TH+)/NeuN + neurons in the substantia nigra instead of the current practice of relying mainly on TH alone as a sole marker of dopaminergic neurons.
Clinical trial design: what new have we learnt since 2019?
In 2019, the workshop concluded that suboptimal trial design around four major areas may have contributed to the fact that the five double-blind phase two studies investigating the neurorestorative effects of GDNF, or its analogue NRTN, had all failed to reach their primary endpoints.25–29
Trial duration
First, any observable benefits from neuroprotective therapies are expected to only produce a slowing in dopaminergic nigrostriatal degeneration and hence only a reduction in rate of clinical decline. Using the MDS-UPDRS part III, the rate of deterioration is estimated to be approximately 2 points per annum 30 and with a 30% reduction in rate of progression against placebo being a likely standard to approve a disease modifying therapy, this translates to a small signal, which is hard to establish in studies that are less than several years in duration—something that is not feasible either financially or from a patient perspective. In contrast, neurorestorative therapies may not only slow progression but also mediate repair and hence show an improvement in clinical scores over baseline. This potential double-win increases the hoped-for effect size and the likelihood of confirming benefit against placebo in neurorestorative trials with implications for the sample size required and length of trial.
In light of this, a recent post hoc analysis of the Bristol GDNF study, tentatively suggested that a clinical response may be seen post GDNF administration, providing some further evidence regarding trial length. In this trial, approximately one third of the study participants receiving repeated GDNF infusions developed OFF state dyskinesias which were phenomenologically similar to those observed at peak L-dopa dose. 31 This suggests that these OFF dyskinesias were related to GDNF-induced heightened levels of endogenous dopamine (although increased serotonin could have played a part). The likelihood of this phenomena emerging increased with each infusion but more commonly only after 9 months 31 which coincided with the time when the double-blind stage ended. Given this, a more optimal trial length may be 18 months, especially if one also tries to hold the standard-of-care drug regime at a steady dose. Furthermore, an 18-month trial rather than one of 9–12 months duration, also makes more sense from a biological standpoint. Whilst terminal sprouting, as consistently suggested in GDNF studies employing 18F-dopa PET, might be expected to occur within weeks to a couple of months, axon regrowth sufficient to restore the nigrostriatal pathway (even if retrograde transport can effectively occur in PD) would take far longer.
Disease subtype and stage
In addition to trial duration, the heterogeneity of PD also suggests more consideration needs to be given to patient selection. 30 In the absence of knowing what increases the likelihood of a GDNF responder versus a non-responder, attempts on how this should be done to enrich for optimal study cohorts is speculative. However, it is generally recognized that disease stage is of critical importance, with the need to go earlier in the disease course when there is more of the remaining nigrostriatal pathway to rescue. While this is a reasonable position to adopt going forward (but see below), it may not apply to all disease modifying agents. As neurorestorative therapies can bring improvements over baseline it may be better to recruit people with mid-stage PD given that having a lower baseline would allow one to capture a greater absolute improvement. Furthermore, regulators have been moving towards an increase in good quality ON time without troublesome dyskinesia as their preferred primary outcome in neurorestorative trials. Such a functional endpoint does have benefits but of course necessitates that participants have reached the stage where they are experiencing motor fluctuations. 32 Maybe setting inclusion criteria similar to those adopted in the EARLY-STIM DBS trial may offer the best chance of confirming efficacy at phase II/III, with the aim of moving earlier only after regulatory approval. 33 The improvement in 18F-dopa PET scans observed in GDNF studies suggests that people 8 years post diagnosis have enough dopamine terminals left to be able to generate a significant PET response across the putamen including the caudal portion. 29 That said, 18F-dopa PET change does not equate to axonal restoration or recovery of a functional pharmacology, which may only be achievable earlier in the disease course. In essence, the jury is out and for the meantime it may be best to keep an open mind about the optimal disease subtype and stage to investigate but with a consensus that earlier in the disease course may offer some advantages.
Sham controls
Placebo effects have been seen in some surgical studies in PD although not all and this needs to be optimally managed throughout the duration of trials. Separately, the development of psychological profiling and imaging methodologies to flag and then exclude those prone to the placebo response are underway. 34 Currently, though, it is unlikely that regulators and sponsors would favor enrolling those with the ‘placebo related pattern” on imaging. In addition, the use of video recordings and blinded independent raters has also shown some merit. 35
Trial end points
The clinical rating scale score used as the primary endpoint in prior neuroregenerative studies has been the MDS-UPDRS part III (or its prior version) in the practically defined OFF state.25–29 To be deemed to have produced a “clinically important difference” (CID) a therapy needs to improve the MDS-UPDRS part III by around 3 points at minimum. 36 However, the motor sub-score has an inter-rater variability of up to 16 points 37 and a relatively low sensitivity to detect change at a patient level. 38 This scale is less sensitive to early-stage disease and displays a marked ‘floor effect’ for most items. 38 Further, the nature of the observed testing situation that the MDS-UDPRS part III requires, means that confounds of observer bias and the ‘Hawthorn Effect’ (where aspects of performance are modified because the person is being observed) are introduced.39–41
An alternative, which enables a more continuous in-home and patient-focused evaluation of PD, is a self-report diary. Diaries such as the Hauser diary have been used in previous GDNF studies to measure ON/OFF fluctuations in motor features as a secondary outcome and more recently the FDA, EMA, MHRA and BfArM have all recommended improvement in good quality ON time without troublesome dyskinesia as the primary outcome measure in future trophic factor trials. However, diaries are subject to recall bias and have poor temporal resolution 42 as well as diary fatigue which reduces compliance with entries. 43 So, whilst diaries afford an insight into someone's subjective severity of PD symptoms, they are felt too burdensome on participants to use over a longer period than four days.
Of late, there has been much enthusiasm for technology-assisted mobility outcome measures44–47 but the concept of digital health is not a new one. However, much of the prior work using such measures have been cross-sectional, done in a laboratory or clinic setting with non-standardized methods and using varying techniques to validate outcomes. Indeed, until very recently, the research activities undertaken by participants with PD have often been scripted and/or part of pre-defined clinical tasks.46,47
Although it is clear that wearables and smartphones show promise as digital tools to measure outcomes in PD, they are unable to add the real-world context of what was actually happening at the time the measurements were being taken. Furthermore, wearables are limited to measuring data from the body part to which they are applied, and smartphones usually require user interaction (therefore technological literacy and the person's active attention during assessment). The use of multiple passive sensors of different modalities deployed in the homes of PwP, overcome some of the limitations of wearables and smartphones. This approach shows promise because it offers functionally relevant, sensitive and reliable clinical measures with continuous monitoring, well placed to better understand the context in which the clinical feature takes place.48–51 Linked to this might be the use of patient reported outcome measures that more accurately reflect the real world problems of PwP. 52
In summary, the type and stage of patient along with the length of any GF trial and measures of end-point efficacy have been refined based on a re-analysis of previous trial data as well as clinical trial developments more generally. Exactly what this looks like in terms of trial design depends to some extent on whether the therapeutic is primarily neuroprotective or neurorestorative.
Is there a stage of disease beyond which GFs cannot work?
Clinical trials of GDNF in PD have failed to reach their primary end-points because, it is often claimed, they are being used at a stage of disease where there is no dopaminergic nigrostriatal pathway to be rescued. However, if this were true, how could the reported increases in 18F-dopa PET signal be seen? 29 As such is it true that beyond a certain stage, the dopaminergic system cannot be rescued? And how exactly does the α-synuclein pathology of PD impact on GF signaling and effects over the disease course?
In order to answer this, we need to better understand the status of GDNF and NRTN signaling in PD. Early studies reported that viral delivery of GDNF to the striatum and/or the SN failed to prevent viral vector expressed A30P mutant 53 or wild-type α-synuclein-induced degeneration. 54 Subsequently it was reported that GDNF signaling was blocked in the rat nigra by down regulation of GDNF signaling receptor Ret expression. 13 This reduced Ret expression in the SN in α-synuclein rodents models is supported by some, 55 but not all studies, 56 but experimental design differences may explain this as may the degree of reduced Ret expression. In this latter respect it has been shown that 50% reductions in Ret has no effect on dopaminergic neurons. 57 The importance of Ret in nigral dopaminergic neurons is underscored by reports showing that intranigral AAV-Ret protected against α-synuclein-induced degeneration in a rat model, 58 and that Ret is critical for the dopaminergic neurotrophic effects of GDNF. 59 Therefore, the status of Ret expression in PD has been an open and important question.
A recent report has directly addressed this question by analyzing the postmortem SN from individuals with no motor deficit (NMD), minimal motor deficits (MMD), and PD. 60 Quantification of Ret in nigral neurons, revealed that neurons with α-synuclein inclusions had lower Ret expression than those without α-synuclein inclusions both in MMD and PD. 59 Furthermore, cellular levels of phosphorylated S6 ribosomal protein (p-rpS6) (an indicator of GDNF signaling)61,62 were reduced in MMD and strongly reduced in the PD groups. 60 There was a strong correlation between Ret and p-rpS6, indicating that Ret may be the primary determinant of p-rpS6 levels, and by extension, GDNF responsiveness, in nigral neurons. 60 It is important to note that although the total number of nigral Ret-immunopositive neurons was reduced in PD, which is expected since dopaminergic nigral neuron loss is pathognomonic of PD, more than 80% of those neurons that remain, continue to express Ret. 60
These findings are further supported by recent data, which showed there was a statistically significant moderate to strong correlation between Ret levels and seven dopaminergic markers (SLC18A2, TH, DDC, SLC6A3, KCNJ6, ALDH1A1, and NR4A2) in controls, which was maintained in PD. This suggests that reduced Ret expression in PD may reflect neuronal loss, rather than downregulation of Ret transcript expression. In support of this, no significant differences in Ret expression in data from laser-captured microdissection (LCM) of individual SN neurons was found, 63 in line with earlier studies.56,59
As cellular levels of p-rpS6 are reduced in PD, 60 it interesting to note that p-rpS6 controls the translation of a subset of mRNAs in a region specific manner in the CNS. 64 Therefore, the p-rpS6 level may determine the level of Ret translation in the SN. This would be consistent with the correlation between Ret and p-rpS6 levels in nigral neurons, 60 and with post-mortem data from a patient who had received AAV2-NRTN (Cere120) to the putamen plus SN bilaterally.65,66 There were more Ret and p-rpS6 neurons in the SN following nigral AAV2-NRTN delivery, suggesting that AAV2-NRTN upregulated Ret expression.65,66 This proposed ‘wind up mechanism’ may lead to a gradual potentiation of GDNF family signaling in nigral neurons that retain sufficient Ret expression. 67 This suggests a scenario that early treatment with GDNF (and related ligands) may be beneficial even in the presence of α-synuclein pathology.
Further data supporting this hypothesis comes from work where primary mouse midbrain cultures exposed to human α-synuclein PFFs, had a significant loss of DA neurons after three weeks, which GDNF prevented. 68 Furthermore, primary cultures of the rat midbrain which were transduced with AAV-GFP or AAV-α-synuclein, cultured for 5 days, and then treated with or without GDNF for a further five days, showed that GDNF prevented α-synuclein-induced reductions in neurite growth. 63 These data suggest that GDNF can signal in cells expressing pathological forms/levels of α-synuclein. This interpretation would also help explain; (i) the case of patients with OFF-state dyskinesias that correlated with GDNF administration 31 and (ii) the report showing greater numbers of nigral Ret and p-rpS6 immunopositive neurons following nigral AAV2-NRTN. 66 α-synuclein may therefore not be the critical determinant of GDNF responsiveness, but rather it may depend on the extent to which Ret expression and signaling is intact. Considering that individuals with MMD still have approximately 55% of their nigral neurons with 85% of them expressing high levels of Ret,60,65,69 then individuals at an earliest stage of PD may benefit most from neurotrophic factor therapy with GDNF family ligands.
Another important criterion for the potential utility of GDNF is a better understanding of whether the dopaminergic neurons in the nigrostriatal system are dead or whether they may be ‘hibernating’ and just cannot be visualized using standard immunohistochemical procedures; the most common approach being staining for TH. Indeed, while α-synuclein has many biological effects on nigral neurons, one underappreciated phenomenon is how α-synuclein downregulates the dopaminergic phenotype in 1) monkey and human normal aging 24 ; 2) in PD 70 ; and 3) in a primate preformed α-synuclein fibril model. 71 Increases in 18F-L-dopa PET in PD subjects following GDNF delivery support this view. 29 Furthermore, there are many nigral neuromelanin positive/TH negative neurons in PD but almost all neuromelanin positive nigral neurons are TH positive nigral neurons in controls. 72 At the transcript level, studies have also shown reduced expression of TH, without a corresponding reduction in NeuN up to 20 weeks post intranigral injection of AAV-α-synuclein in rat models.55,73 These data, taken together support the concept that PD nigral neurons may not be able to be seen with TH, and perhaps other dopaminergic markers (e.g., dopamine transporter (DAT)) may be more useful- which further supports the view that a consensus on how best to visualize true dopamine cell loss is needed (see also above discussion).
In summary, there is a complex interplay between Ret signaling, alpha synuclein pathology, TH expression and responsiveness to GDNF and related GF therapies but on balance targeting early stage patients may be the sensible first step in future trials of these agents.
Do we know how to optimally deliver GDNF and related GFs to the PD striatum?
A consensus opinion is emerging that precise, targeted distribution to the putamen of either recombinant GDNF protein or AAV vectors carrying the GDNF transgene will provide optimal therapeutic opportunities for mitigating the clinical features of PD. Both therapeutic formulations have been safely and effectively delivered within the putamen over time using advanced applications of convection enhanced delivery (CED).4,29,74,75 CED provides a pressure-driven bulk flow of the infusate containing either protein or vector, and optimizes the localized extracellular volumetric distribution of either agent within the target brain parenchyma. 76 In its current form, CED operates using a microinfusion pump-dependent drug distribution approach, over a period of hours, via uniquely-designed catheters/cannulae, precisely-placed and monitored using magnetic resonance imaging (MRI) during or immediately following the infusion. 77 Using such focal intraparenchymal brain delivery to the putamen has avoided clinical side effects as were seen following more widespread GDNF exposure within the CNS. 25
The clinical effect size of GDNF therapy within the striatum (primarily putamen) largely depends on the availability and distribution of GDNF protein as well as its transport to the substantia nigra pars compacta (SNpc).78–80 Anatomical pathway interconnections between the putamen and substantia nigra are important for all types of GDNF therapies. Intraputamenally delivered GDNF protein is dependent on synaptic uptake and retrograde transport from the putamenal dopaminergic terminals to the SNpc somata via the retrograde nigrostriatal dopaminergic transport pathway.81,82 As the pathway progressively degenerates and dopaminergic neurons are lost with advancing stages of PD, 72 the clinical effect is likely to diminish, along with the retrograde transport. With the GDNF transgene-carrying AAV2 vector (AAV2-GDNF) delivered to putamen, however, there are two options for transport to the substantia nigra. First, there is transduction of putamenal neurons that locally produce and secrete GDNF protein, helping sustain putamenal dopaminergic terminals and their SNpc neurons (via retrograde transport). Second, the same putamenal infusion allows AAV2-GDNF to undergo anterograde transport via the largely preserved GABAergic striatonigral pathway to the substantia nigra pars reticulata (SNpr), where it transduces resident neurons, leading to local production and secretion of GDNF, which can then resuscitate and help maintain the adjacent SNpc dopaminergic neurons. 83
Previous human PD trials have successfully tested the safety and tolerability of nigral transduction via direct delivery of AAV2-NRTN, although delivered volumes were limited to 30 μL per substantia nigra. 84 Initial nigral/ventral tegmental area (VTA) CED approaches confirmed the safety and efficacy of AAV2 vectors containing the human aromatic amino acid decarboxylase (hAADC) transgene, assessed in children suffering with AADC deficiency (AADC-d). 85 Using separate trajectories to the midbrain targets, the reported first seven AADC-d participants were infused with 50 μL per substantia nigra (n = 13) and 30 μL per VTA (n = 14). Current approaches for the treatment of AADC-d include bilateral single trajectories passing between the VTA and SNpc and distributing CED infusion volumes (Vi) of up to 300 μL per side to encompass both structures. 86 Although yet to be tested in PD patients, AAV2-GDNF infusions are considered safe using similar nigral or nigral/VTA approaches and volumes as previously shown with AAV2-NRTN and AAV2-AADC, should a need for such an option arise.
Putamenal Vi provided by CED have increased over time from less than 100 μL per putamen to the current standard of up to 1800 μL. 87 In the majority of recent human putamenal CED infusions evaluated, for each 100 µL of Vi delivered within the target, the corresponding volume of distribution (Vd), assessed via the visualized gadolinium co-infusion on the intraoperative MRI, varies between 200 μL to 300 μL, thereby providing a Vd/Vi ratio of 2:1 to 3:1. 88 With such Vd/Vi ratios for the putamen, and a consistent use of Vi in the range of 1000 μL to 1800 μL per putamen, volumetric putamenal coverage of between 50% to 80% is anticipated, with corresponding improvements in efficacy while maintaining safety and tolerability. Evolution of infusion methods that parallel the long axis of the putamen have resulted in larger Vi for both protein infusions and gene therapies. An occipitoparietal putamenal approach was tested in two Phase 2 clinical studies of GDNF protein infusions,29,74 reproducibly yielding mean putamenal volumetric coverages of 47.8% to 57.2% using 2 catheters to deliver a total infusion volume of 800 µL per putamen. For MRI-monitored AAV2 gene therapy approaches, an occipitoparietal “infuse-as-you-go” technique89,90 has been developed to optimize coverage of the putamen's non-spherical shape while continuing to minimize distribution outside the target.
As the two studies testing intermittent intraputamenal delivery of GDNF protein have not yielded any safety concerns, and the total dose given per 4 weeks in these studies (240 µg) was substantially smaller than in prior continuous dosing intraputamenal delivery studies (approximately 800–2400 µg),26,29,74,91–93 it is suggested that raising the intermittent dose closer to the lower end of the dose range tested with continuous delivery is justified. Due to practical restrictions on catheter number, infusion frequency and Vi, this can be most easily achieved via increasing the GDNF protein dose concentration from 0.2 µg/µL to 0.6 µg/µL, thereby raising the total dose given per 4 weeks from 240 µg to 720 µg. Justification for such an increase is also provided by the absence of local or remote lesions in a 9-month toxicity study that tested the intermittent intraputamenal delivery of GDNF at 0.67 µg/µL in rhesus monkeys. 94
Human GDNF gene therapy trials to date have tested batches of the same AAV2-GDNF production run. In a recent Phase 1 trial including 13 subjects with advanced PD, three vector concentrations (9.9E + 10, 3.3E + 11, and 9.9E + 11 vg/mL) were tested with a consistent Vi of 450 µL per putamen, yielding maximal putamenal doses of 4.5E + 10 vg (n = 6), 1.5E + 11 vg (n = 6), and 4.5E + 11 vg (n = 1), respectively. 4 In the most recent Phase 1b trial including participants with mild (n = 6) and moderate (n = 5) stage PD, a single vector concentration (3.3E + 12 vg/mL) was used with a Vi of up to 1800µL per putamen, yielding a maximal putamenal dose of 5.94E + 12 vg (NCT04167540; unpublished data). The Phase 1 study delivered putamenal doses using bifrontal infusion approaches, with at least two separate trajectories per putamen. The Phase 1b study used a single occipitoparietal trajectory per putamen in conjunction with the “infuse-as-you-go” technique. Both studies showed a favorable safety profile for AAV2-GDNF up to 5 years post treatment (unpublished data).4,75
In summary, the optimal dose and delivery approach for infusing GDNF or AAV2-GDNF into the striatum is being addressed, although it is still under investigation whether this can be further improved upon. What is clear is that many of the earlier GF trials used suboptimal methods and volumes/doses for delivering their therapeutic agent to the PD brain.
Is it better to develop GDNF like GF therapies in a regulatable fashion?
The safety profile of GDNF has generally been very good, but there were early signs of cerebellar toxicity at high doses with continuous intraputamenal delivery in primates 95 and Lhermitte phenomena were often reported in the early days of GDNF intraventricular protein delivery in PD patients. 25 Furthermore, systemic delivery of GDNF does show some toxic side effects.96,97 Thus, the possibility remains for side effects with the continual delivery of GDNF directly to the brain using AAV or cells engineered to stably secrete GDNF.98,99 Thus, having more control on the delivery of this factor would offer some advantages and interestingly there are now exciting alternatives involving regulated promotor systems to drive controlled GDNF expression delivered using these gene and cell systems. The mainstay of these has been doxycycline (DOX) induction elements in the promoter of the gene that has been used in many in vitro and in vivo studies, to turn “on” or “off” the production of GDNF.100,101 DOX, along with similar compounds, has been shown to efficiently switch on and off genes in transplanted neural progenitor cells in the rodent brain.102,103 While very efficient in vitro and in vivo, there are safety questions about long-term administration of DOX to patients at doses needed to control expression, the immune stimulatory effects of the reporter system and the leakiness of the regulatory system. 104
To deal with these concerns, newer technologies include a remarkable system Xon that uses alternative splicing systems to precisely regulate gene expression using orally bioavailable drugs, some of which have already been in clinical trials such as Branaplam. 105 This system inserts the alternative splicing mechanism into cells, which then only make the protein in response to Branaplam administration. The drug itself has been used by Novartis for driving alternative splicing of specific proteins in two clinical trials (for SMA and Huntington's disease). However, the trials were recently stopped due to side effects of Branaplam that included peripheral neuropathies. This attests to the complexity of driving GDNF expression using inducible systems, but the ability to do this remains an active area of research in the gene therapy field.
In summary, while regulating the expression of GDNF post-delivery would be useful, this is currently not thought to be needed or possible clinically given the safety profile of this agent to date.
Is GDNF still the right molecule for repairing the dopaminergic nigrostriatal pathway in PD?
Given all the above controversy around whether GDNF can work in the context of α-synuclein pathology, attention has been directed to other Ret-independent neurotrophic factors, either when administered alone, or when combined with GDNF family ligands to enhance efficacy.
Bone morphogenetic proteins (BMPs), a subgroup of the TGB-β superfamily (which comprise the BMPs and growth/differentiation factors (GDFs)), as well as GDNF family ligands, are secreted growth factors which play important roles in embryogenesis, skeletal development and dopaminergic neurogenesis.106–108 BMPs bind to BMP receptors (BMPRs), which activate intracellular Smad1, which translocates to the nucleus to alter gene transcription.109,110 BMPR2 is expressed on dopaminergic neurons in the adult rat SN, 111 and BMPR expression is higher in dopaminergic neurons in the SNpc compared to those in the VTA. 112 Studies have shown that GDF5 111 and BMP2 113 promote dopaminergic neurite growth through a Smad1-dependent mechanism, and more recently it has been shown that target-derived BMP2/BMP6 regulates nigrostriatal dopaminergic synapse development. 112
Two recent reports have investigated the therapeutic potential of viral delivery of BMPs in α-synuclein rodent models of PD.55,114 The first study employed unilateral intranigral injections of AAV-α-synuclein, along with AAV-GDF5 or a null control. 112 At 20 weeks post-surgery, there was a significant reduction in nigral dopaminergic neurons and in striatal dopaminergic innervation in the control group, which was not seen in the AAV-GDF5 group, coupled with similar changes in striatal dopamine levels. 55 However, it is important to consider whether this reflects neurorestoration of phenotype, or a neuroprotective effect of AAV-GDF5. This study also found that α-synuclein overexpression did not affect the expression of transcripts for Bmpr2, Bmpr1b, and Smad1, whereas Ret expression was significantly reduced. 55 This suggests that BMP receptor expression is maintained in the presence of α-synuclein overexpression. In agreement, another study reported that lentiviral intranigral delivery of BMP5/7 prevented A53T-α-synuclein-induced loss of dopaminergic neurons (as well as a reduction in total numbers of NeuN + neurons) and prevented motor impairments and associated gliosis in a mouse model of PD, 114 suggesting a neuroprotective effect of BMP5/7 overexpression in this setting. This study also demonstrated that a loss of BMP/SMAD signaling led to α-synuclein accumulation in nigral dopaminergic neurons, while conversely BMP5/7 overexpression reduced α-synuclein levels in the SN. 114 Interestingly, the study also reported increased levels of the inhibitory Smad6, which inhibits BMP-Smad signaling, in postmortem PD nigral samples. Whether the neuroprotective effects of BMP5/7 are mediated through, or are independent of, such reductions in α-synuclein remains an open question.
In summary, all of this work suggests that there may be other GFs that could be considered for use in PD in order to avoid these issues around changes in Ret expression and signaling associated with GDNF.
The need to move beyond focal neurorestoration of the dopaminergic system: a shift to more systemic GF like agents?
Intracranial delivery of growth factors or viral vectors or cells expressing GFs has several advantages. Firstly, the delivery will be local and therefore should minimize off target side effects. Secondly, delivery of proteins directly to the brain should reduce the likelihood of any systemic immune response. Finally, the GFs should be properly folded and carry native posttranslational modifications, making them indistinguishable from endogenous GFs. However, intracranial delivery has limitations. Surgery is risky and expensive, and GF protein-based drugs are often limited in their diffusion across the brain. These risks mean that ethical committees will only allow the treatment of later stage patients, which may not be the optimal group for rescuing the dopaminergic pathway (see above). For example, in the last GDNF phase 2 trial, patients had a mean disease duration from first symptom of 10.9+/- 5.3 years 29 and in the CDNF trial of 10.8+/-2.7 years. 2 In addition, non-motor symptoms in PD significantly affect the quality of life of the patients and degeneration and loss of neurons outside the dopaminergic nigrostriatal pathway, and in the periphery play an important role in the pathogenesis of these features. Intracranially delivered GFs cannot reach these sites of pathology and thus treat these aspects of PD.
In addition to the problem related to intracranial delivery, some of the limitations are related to the GF proteins themselves. In this respect, biologically active CDNF peptides that can pass through the blood-brain barrier (BBB) have now been successfully made and tested in vitro and in animal models of PD.115,116 In particular, a modified peptide (Class A peptidomimetic) derived from the C-terminal domain of human CDNF protein 116 has recently been developed. 115 This compound, called HER-096, has superior metabolic stability over native L-peptides and has been shown to cross the BBB in various animal species. 115 This has led to a Phase 1a first-in-human study in healthy volunteers with subcutaneously administered HER-096, which was started in April 2023; the topline data was released at the end of October 2023 (ClinicalTrials.gov ID NCT05915247). 117
Another approach is to develop small molecule chemical compounds that mimic the action of GFs and have better pharmacokinetic and distribution properties than classic GFs. GDNF mimetics have been developed which have been shown to protect DA neurons in culture as well as in animal models of PD.118,119 Likewise, BDNF mimetics have demonstrated some efficacy in animal models of PD. 120 Finally, small molecules that regulate the expression of endogenous GFs are a very promising approach and in this respect rasagiline has been shown to upregulate endogenous levels of GDNF and BDNF, 121 but this line of research has not very extensively pursued.
In summary, small molecule drugs that recruit endogenous GF systems to treat PD have many benefits. These compounds have better PK properties compared to GF proteins, meaning they diffuse better, have a longer half-life and most importantly they reach many brain areas as well as peripheral sites of pathology, allowing for the treatment of non-motor symptoms. In addition, as these drugs can be delivered systemically, it is possible to start treatment soon after PD diagnosis when more DA neurons are still alive with the added advantage that they could be trialed more widely and not just in specialist neurosurgical centers. However, peptides and small molecule drug candidates have some limitations. For instance, when they reach other brain areas and peripheral tissues, there is higher risk for side effects as well as inducing a host immune response especially with peptide therapies.
Moving beyond GDNF to novel GF agents such as CDNF?
CDNF 122 and the related growth factor mesencephalic astrocyte-derived neurotrophic factor (MANF) 123 form a novel family of growth factors with neurotrophic factor-like activities. CDNF and MANF are monomeric non-glycosylated proteins of 161 amino acids and 158 amino acids, respectively. Although both proteins have the N-terminal signal sequence, they are mostly located on the luminal side of the endoplasmic reticulum (ER) and are secreted at times of cellular stress through a mechanism involving calcium depletion. Compared to other neurotrophic factors, CDNF and MANF have unique structures.124–128 They have eight conserved cysteine residues with identical spacing and both proteins have an N-terminal saposin-like domain and a C-terminal SAP-like domain.124,125 They also have a different mode of action compared with other known GFs. Firstly, they promote the survival of neurons and induce neurite outgrowth by a dual mechanism. They mostly act inside the cells regulating their receptors on the ER membrane, but they can also act as extracellular proteins.129,130 The neuroprotective effects of CDNF and MANF are linked to the unfolded protein response (UPR) pathway, which operates in the ER. MANF can bind to, and regulate, the activity of the IRE1α receptor on the ER membrane protecting DA neurons in vitro and in vivo 130 while CDNF binds to ER chaperones, with GRP78 playing a particular role. 131 CDNF can also rescue motor neurons in an animal model of motor neuron disease (ALS). 131 MANF can also bind to and activate plasma membrane receptor neuroplastin (NPTN), regulating the inflammatory response and apoptosis of cells. 132 Secondly, they have very little or no effect on naïve cells,122,133 likely because in homeostatic conditions the receptor is occupied by another ligand. 130 Thirdly, they can regulate the unfolded protein response (UPR) and reduce protein aggregation in the cells.18,125,134–136 CDNF can prevent α-synuclein entry into neurons, reduce its aggregation, and alleviate motor deficits in the mouse α-synuclein PFF model of PD. 18 Finally, CDNF can also reduce neuroinflammation by decreasing the synthesis and secretion of pro-inflammatory cytokines.137,138 In rodent MPTP and 6-OHDA models and non-human primate 6-OHDA models of PD, CDNF, either delivered as a protein or gene therapy, can protect and regenerate DA neurons and significantly improve motor function.18,121,133,139–141 On the basis of this pre-clinical data, CDNF was recently tested in a phase I clinical trial. The primary objective of this clinical trial was to assess the safety and tolerability of monthly intraputamenal CDNF infusions in PD patients. The second objective was to investigate the safety of the drug delivery system (by Renishaw Neuro Solutions). The CDNF phase I study in moderately advanced PD patients met the primary endpoint because the drug-related adverse events were either mild to moderate in terms of severity with no difference between placebo and treatment groups. Even though later-stage patients with a significant loss of midbrain DA neurons were treated, positive CDNF effects were still seen in some patients. 2 Developing this agent to be delivered systemically using a smaller BBB penetrant version of it (HER-096) is now underway (see above).
In summary, there is now great interest in exploring a new class of GF in PD, CDNF, that may have an array of effects around different postulated pathogenic pathways in PD, which may treat all aspects of PD as it could be given systemically.
Is there merit in using GDNF and related GFs with pluripotent stem cells?
A number of first-in-human studies have now commenced with pluripotent stem cell drug products differentiated into dopaminergic precursors for use in people with PD. As these effort continue, next-generation products are expected to emerge. The expression of GDNF or other GFs from engrafted A9 dopaminergic neurons represents a distinct approach. This approach involves transplanting cell therapy products derived from embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), which mediate circuit restoration and provide temporally controlled expression of GFs, such as GDNF. This dual approach supports and rejuvenates endogenous dopaminergic cells that are dysfunctional but which are still capable of responding to trophic support (Figure 1). A combined cell transplantation and GF delivery approach has the advantage that the cell therapy will replace lost neurons, which provides the structural foundation for rebuilding neural circuits while the GFs promote survival of existing neurons. This approach will need to consider a number of factors to achieve a potentially more potent cellular drug product that would be amenable to a broader range of patients.
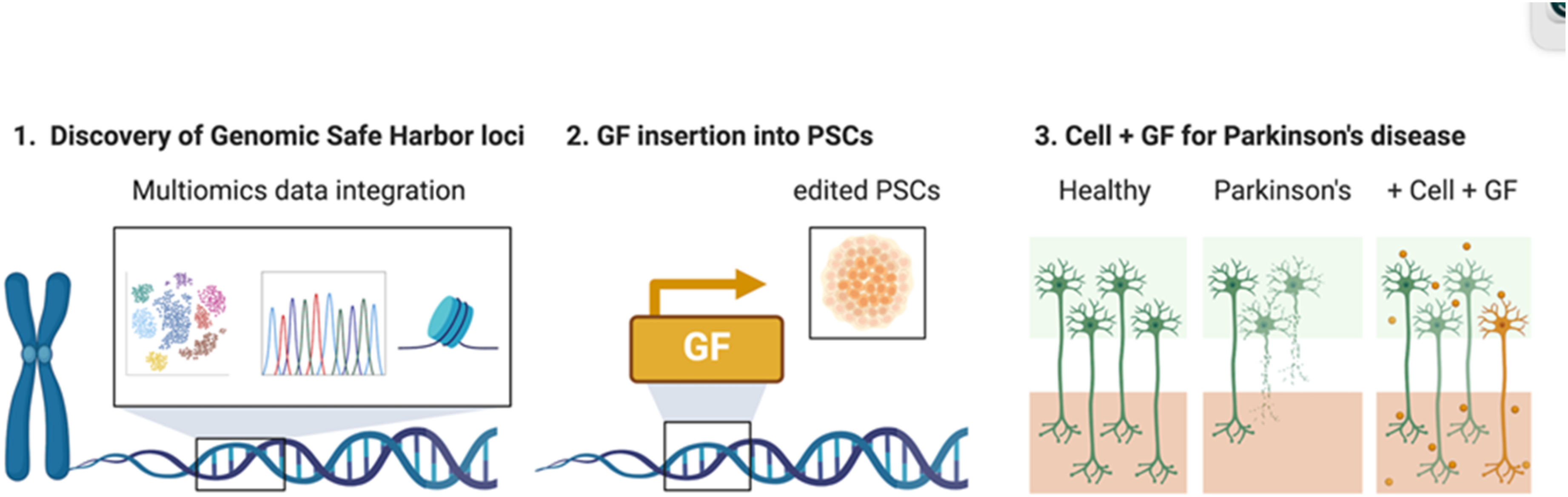
Combination of cell replacement therapy and growth factor delivery as a therapy paradigm for Parkinson's disease. This paradigm involves (1) Discovery of Genomic Safe Harbor (GSH) loci through multiomics data integration, ensuring that the selected loci are suitable for stable and safe gene insertion. (2) Using precise genome editing tools, growth factor (GF) genes are knocked into the identified GHS loci in substrate pluripotent stem cells (PSC). (3) The final gene-modified cellular product expressing growth factor (Cell + GF) is predicted to reconstitute the lost neural circuitry (orange cells) and promotes survival of existing neurons by secreting GF (orange circles).
First, the choice of which GF to express is based on the intended target (e.g., A9 dopaminergic neurons), the known engagement with cognate receptors, and the feasibility of pharmacodynamic readouts that allow differentiation from the cell product lacking additional GF function (see above discussion).
Second, it is vital to determine the optimal timing of GF gene expression after transplantation. The temporal pattern of expression should not compromise the neurobiological differentiation of pluripotent cells to mature post-mitotic A9 neurons. Limited data from the rat Parkinsonian model suggests that using AAV-GDNF to transduce the denervated striatum in relation to graft placement timing affects both the graft-derived A9 dopaminergic survival and host innervation by engrafted post-mitotic dopaminergic neurons differentially. 142 These observations suggest that delaying GDNF expression relative to graft placement results in benefits, as evidenced by behavioral recovery in rat models. Accordingly, the design of the expression cassette, inclusive of pertinent cis elements and other features, should be considered to achieve proper transactivation of the GF gene and possibly sustained expression to achieve an autocrine/paracrine trophic stimulus to both grafted and endogenous dopaminergic neurons.
Third, attention to the method of engineering the cellular substrate is important to avoid genomic modifications that may alter the intended cellular phenotype. Viral vectors are the current standard for delivering therapeutic genes in cell and gene therapies (CGTs). However, the random integration of viral vectors raises safety concerns in clinical applications. An alternative approach is through identifying the optimal Genomic Safe Harbor (GSH) loci that are suitable for editing transcriptional cassettes containing the therapeutic gene product.143,144 This approach mitigates the risk of de novo tumorigenesis by deliberately avoiding the placement of therapeutic genes adjacent to proto-oncogenes and other important genomic elements, which may occur inadvertently with viral insertion or off-target editing. Identification of optimal GSH sites requires multiomics data integration (Figure 1) and the selection of editing methods to achieve the placement of the transcriptional cassette into the best GSHs that meet temporal expression criteria, avoid potential oncogenic activation, and do not adversely impact on the differentiation of the cellular drug product.
Fourth, establishing criteria for the in vivo performance of such gene-modified cellular products that address potency, retention of cellular identify and sustained non-clinical benefit in optimal models of PD is required. Potency involves ensuring that the genetic modification results in the desired biological activity, such as dopamine production, with optimal dosing that maximizes therapeutic effects while minimizing side effects. Retention of cellular identity is crucial for maintaining the intended phenotype, which requires monitoring phenotypic stability and genetic integrity through molecular markers and functional assays. Sustained non-clinical benefit demands long-term efficacy studies in relevant animal models, assessing motor function, neuroprotection, behavioral tests and histological analyses. Implementing these criteria involves designing comprehensive preclinical studies aligned with regulatory guidelines.
In summary, there is great merit in considering combined therapeutic approaches in PD using both dopaminergic cell replacement from stem cell sources combined with GF delivery. Exactly how this can be done optimally is unknown, not least because the data showing that stem cell derived dopamine cell transplants work reliably and robustly in people with Parkinson's is still to be shown.
Is there merit in using GDNF and related GFs with non neuronal cells?
It is becoming clear that in addition to dopamine fiber loss, there is also inflammation in the striatum of PD patients. Furthermore, there is also clear astrocyte dysfunction that may compound or even contribute to dopaminergic neuronal degeneration. 145 Neural progenitor cells that give rise to astrocytes following transplantation to the striatum can be genetically engineered to produce GDNF or CDNF and thus represent a combined cell and growth factor approach to treat PD. 101 The new healthy astrocytes may dampen inflammation and release a range of growth factors to stabilize the inflamed environment. Furthermore, recent data suggests that new glial progenitor cells may be able to replace sick or dying glia in the adult striatum 146 and thus could both reduce the inflammatory milieu and provide a new support cell that also releases GDNF or CDNF.
In summary, delivering GDNF using engineered PSC or other progenitor cells as a monotherapy may offer some advantages, but currently the benefits of this require further exploration.
How competitive will all these GF therapies be?
One of the key questions that will ultimately need to be addressed by any GF therapy is whether it can compete against what is in clinic already as well as what may be coming down the line. 147 In this regard, it depends what the therapy is trying to rescue/restore. If it is the dopaminergic nigrostriatal pathway then this therapy will have to offer some advantage over current dopaminergic treatments for PD including infusions of dopamimetic agents (e.g., Apomorphine infusions; DuoDopa®). Obviously, GFs are trying to regrow or preserve this pathway and clearly if they do this to the point that the patient has sufficient innervation of the striatum (>20–30% of normal), then this will mean they do not need to take any dopaminergic drugs. Such an intervention would offer a major competitive advantage over these other more invasive continuous dopaminergic agents. However, if they cannot achieve this and only partially reduce the need for the PwP to take dopamine drugs then it is less clear whether this is sufficient to make them competitive. This may become more of an issue if experimental approaches using dopamine cell or gene therapies prove effective—therapies that are now going to first in human trials in PwP and which have the advantage of bypassing the need to keep alive a diseased dying network.
If on the other hand, the GF works through a cell rescue mechanism and can be given systemically then this puts this therapy into a category of its own, given we have no disease-modifying therapies for PD. Thus it would then compete with all other agents being trialed for this purpose (see 148 ).
In summary, the competitiveness of GF therapies for PD depends on their mode of action, the size of the effect they produce as well as the complications of their administration. Currently they remain competitive although whether this continues to be the case will depend on the next trial results as well as the final cost of the approved therapy if shown to be effective.
Conclusion
There are number of conclusions that can be drawn from this recent workshop and with this a number of next steps have been identified which are summarized below and in Table 1
Preclinical modelling of GF has moved away from neurotoxin to alpha synuclein models. However, it is still not clear whether all such models are equivalent and so further work is needed to generate a consensus on the best model to be adopted prospectively by all groups. In addition, the need to define whether the GF is neurorestorative or neuroprotective is paramount as this has implications for trial design (see below) as well as preclinical modelling. In this last respect, better defining the integrity of the nigrostriatal is urgently needed with a move away from simply using TH immunochemistry of the nigrostriatal system. Linked to this has been the controversy as to whether alpha synuclein impacts on Ret signaling and thus the ability of dopaminergic cells to respond to GDNF and similar GFs. Although not fully resolved, it does seem that alpha synuclein pathology does not mean dopaminergic neurons cannot respond to such GFs. Most of the current work on GFs in PD is looking at a new generation of therapies that have already gone to clinical trials including AAV2-GDNF and CDNF. However, in this last respect there has been a move away to systemically delivered agents that work in a similar way to CDNF (HER-096) which offers many advantages. In addition, new GFs are also still being explored (e.g., GDF 15) and should continue to be, given the clinical data from trials with GDNF and CDNF to date. Aspects of trial design have evolved but are still not full resolved around the optimal subtype of PD patient to enroll in these trials, the duration of the study and the degree of blinding and the best trial end points. Linked to this is the vital need to have patient input on all this including patient related outcomes that are meaningful to the PwP; this work needs further exploring including with the regulatory authorities. Linked to this has been the question of disease stage and whether there is a stage of PD beyond which this therapy cannot work due to loss of the dopaminergic network and/or a degree of alpha synuclein pathology that would prevent the cells from responding to the GF (esp. GDNF). This now seems to be less of a concern, but overall it is recommended that earlier stage patients are recommended for trials of GFs given that they have more of a dopaminergic system to work with in terms of neurorestoration and/or neuroprotection. Optimal intracranial delivery of GFs to the putamen in PwP is being resolved using CED devices capable of infusing large volumes of agent along the axis of the striatum. Although the optimal dose still remains somewhat unresolved, this seems less of an issue than it was in 2019. However, there is interesting new work around delivering GDNF using either regulatable forms of it or delivering it using a cell transplant with the hope that the grafted cells may modulate the host environment in non GF ways. This work is certainly worth exploring further preclinically especially given that stem cell transplants of GDNF are in trials in motor neuron disease, and stem cell derived dopamine therapies are in the clinic for PD. The competitive nature of these therapies going forward depends on resolving much of the above as well as the success of other innovative approaches to treating PD including both repair using cell therapies and disease modification using a range of approaches. However scoping out the likely cost of any GF therapy, its clinical market and how this compares to other regenerative therapies that are now going to clinic should be undertaken, and may already be well developed in companies pursuing such approaches (e.g., AskBio).
Overall, the workshop covered much new ground around GDNF and related GFs with more optimism and hope that this therapeutic approach has merit, which is borne out by the recent announcement of a new GDNF gene therapy trial in PD as well as the possibility of a new systemically delivered CDNF trial.
Footnotes
Acknowledgments
We would like to thank Aleksandra Pilcicka at Cure Parkinson's for all of her patience and work in helping bring this multi author contribution together into one coherent review. We would also like to thank the Cure Parkinson's charity who helped organise this workshop in conjunction with Michael J. Fox Foundation and Parkinson's UK.
ORCID iDs
Funding
Cure Parkinson's solely funded this workshop, but the views expressed were those of the assembled committee given independently of this funding.
Declaration of conflicting interests
RAB receives grants from Cure Parkinson's, MJ Fox Foundation/ASAP, EU, Rosetrees Trust and the MRC. He has paid consultancies with Aspen Neuroscience, Novo Nordisk, Bluerock Therapeutics, Bayer, UCB, Treefrog therapeutics, Amphista and LifeArc. He received royalties from Wiley and Springer-Nature. RAB is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
MS receives grants from Cure Parkinson's, EU, Sigrid Juselius Foundation and Finnish Research Foundation. He is the shareholder in Herantis Pharma and GeneCode companies.
CS receives funding from Cedars-Sinai Board of Governors Regenerative Medicine Institute. He has a paid consultancy with Coya Therapeutics.
CM receives funding from the Engineering and Physical Sciences Research Council.
AW receives funding from the Engineering and Physical Sciences Research Council. He has a paid consultancy with Abbvie Inc.
MSF is a paid employee of AskBio.
ML works as a paid consultant for AskBio.
KSB receives grants from NIH, he has paid consultancies from AskBio, Scribe Therapeutics and AviadoBio.
BF is a full-time employee of The Michael J. Fox Foundation for Parkinson's Research which funds and facilitates research and therapy development for people with Parkinson's disease.
AR is a director of Parkinson's UK, which has provided funding for, and holds a financial interest in, the development of a CDNF-related compound by Herantis Pharma PLC for the treatment of Parkinson's.
JHK receives grants from the Michael J. Fox Foundation, ASAP, and the NIH. He has paid consultancies with Kenai therapeutics, ABBVIE Inc., and CureVC.
ELL receives funding from Cure Parkinson's and Parkinson's UK, and is supported by Llywodraeth Cymru through the Health and Care Research Wales (UA05)-funded BRAIN Unit.
HJH is an employee and a shareholder of Herantis Pharma PLC, and an inventor in a patent related to HER-096.
AS and GOK receive funding from Cure Parkinson's, Science Foundation Ireland and the Irish Research Council.
VY holds equity in Kenai Therapeutics.
HJF is co-founder and holds equity in Kenai Therapeutics. He is the PI of the grant from the California Institute for Regenerative Medicine. He is also co-investigator of a grant from the Michael J. Fox Foundation.