
Abstract
Background
Reducing nigrostriatal iron overload reduces neuronal loss in Parkinson's disease (PD) models.
Objective
Examine the safety and efficacy of deferiprone in reducing motor disability progression in dopaminergic-treated and treatment-naïve patients with early-stage PD.
Methods
Two phase II, multicenter studies, SKY and EMBARK, enrolled patients diagnosed with early PD (<3 years from screening). In SKY, patients on stable dopaminergic therapy were randomized 1:1 to one of four dosage (or placebo-matching) cohorts (300, 600, 900, 1200 mg twice daily [BID]) for 9 months. EMBARK enrolled patients on stable dopaminergic therapy or treatment-naïve patients and received 15 mg/kg BID. For both studies, the primary outcome was the change from baseline to month 9 in motor examination score (Movement Disorder Society-Unified Parkinson's Disease Rating Scale [MDS-UPDRS] Part III). ClinicalTrials.gov: NCT02728843; ANZCTR: ACTRN12617001578392.
Results
Overall, 140 patients were randomized in SKY (28 per cohort). Thirty-six patients enrolled in EMBARK (27 dopaminergic-treated; 9 treatment-naïve). In the SKY study, all doses showed the same worsening as the placebo group, with the exception of the 600 mg dose, which was associated with non-significant reductions in MDS-UPDRS Part III least-squares mean (LSM) between baseline and 9 months (−2–8 points versus placebo). In EMBARK, LSM (SE) changes from baseline in MDS-UPDRS Part III were nonsignificant (–1.6 [1.7]) and significant (8.3 [3.9]) for dopaminergic-treated and treatment-naïve patients, respectively, the latter indicating disease worsening. Adverse events possibly related to deferiprone were reported in 35.7%–88.9% across all deferiprone groups vs. 42.9% for placebo.
Conclusions
SKY and EMBARK studies indicate that deferiprone combined with L-dopa does not provide significant motor function benefit, while the absence of L-dopa treatment worsens symptoms.
Plain language summary
Plain language summary: Parkinson's disease is an age-related brain condition that can lead to problems with movement, muscle cramps, mental health, and pain. People with Parkinson's disease also struggle with activities of daily living that can reduce their well-being. Researchers have found that some people with Parkinson's disease have high levels of iron in their brain. Currently, we do not know if removing iron from the brain can help people with Parkinson's disease. In this study, we examined if a medicine that removes iron from the brain could improve symptoms of Parkinson's disease. This medicine is called deferiprone. Our study included 176 people with Parkinson's disease. People were then divided into 2 groups. In the first group, people were already being treated for Parkinson's disease and then were given deferiprone for 9 months. We found that a low or high dose of deferiprone did not improve disease symptoms. In the second group, people taking no other medication for their Parkinson's disease were given deferiprone for 9 months. In this group, deferiprone worsened Parkinson's symptoms. There are currently no arguments in favor of using deferiprone in Parkinson's disease. Deferiprone without L-dopa worsens symptoms. Research could potentially evaluate low doses of iron chelator in combination with L-dopa over the long term and in large numbers of people.
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by motor and nonmotor symptoms, affecting about 1% of the population aged 60 years or older.1,2 Hallmarks of PD include the loss of dopaminergic neurons in the substantia nigra, Lewy pathology, and iron accumulation in the nigrostriatal pathway.3–6 Excessive free iron can cause oxidative damage and iron-related cell death (ferroptosis),4,7,8 leading to localized neuronal death 9 associated with loss in movement control. 3 Although dopaminergic treatments can reduce symptom severity, there is currently no neuroprotective agent to reduce PD progression. Iron chelation therapies that reduce iron levels only in key brain regions in early-stage PD could potentially slow disease progression.4,6,7,10
Deferiprone is an iron chelator approved for treating systemic iron overload. Deferiprone is associated with specific pharmacological properties that may reduce iron-dependent free radicals and allow for conservative iron chelation.11–13 Deferiprone crosses the blood-brain barrier14,15 and can effectively remove excess intracellular iron from the cell by binding labile iron and creating an iron-chelator complex. 15 Previous clinical studies in PD applied this iron chelation strategy and demonstrated improvement in motor function with oral liquid deferiprone at 10 or 15 mg/kg BID for 6 months among patients with early-stage PD on dopaminergic treatments.10,16 However, a recent trial with newly diagnosed, drug-naïve patients reported worsening parkinsonism with 15 mg/kg BID compared with placebo. 17 Thus, questions remain regarding which patients could potentially benefit from deferiprone and at what dose.
The Study of Parkinson's Early Stage with Deferiprone (SKY) and the Efficacy and Safety of Deferiprone in Treatment-Naïve and Non-Treatment-Naïve Patients with Parkinson's Disease (EMBARK) studies examined the efficacy and safety of deferiprone across 9 months of treatment in patients with early-stage PD. The objective of SKY was to establish the fixed dose of deferiprone tablets (twice daily) for optimal clinical efficacy and safety. The objective of the EMBARK study was to determine whether response to deferiprone differed between patients on stable dopaminergic therapy and those who are treatment naive.
Methods
Study design
The SKY study was a phase II, multicenter, randomized, double-blind, placebo-controlled, dose-ranging study in patients with PD. The EMBARK study was a multicenter, open-label, single-arm study in patients with a diagnosis of idiopathic PD. Both studies comprised a 30-day screening period, followed by a 9-month treatment period, and ended with a safety follow-up at month 10. Both studies were conducted according to Good Clinical Practice guidelines of the International Conference on Harmonization and the ethical principles of the Declaration of Helsinki. Ethics committees of each country approved the conduct of the studies. An independent Data Safety Monitoring Board (DSMB) was established to monitor the safety of patients during their respective studies. Members of the DSMB were responsible for overseeing the conduct of the trial and were empowered to recommend stopping the trial if, in their judgment, continuation was not ethically acceptable on the grounds of safety. The DSMB conducted interim review meetings, during which a thorough safety review of the studies were conducted in consultation with the sponsor. The safety evaluation included clinical assessment of adverse events (AEs), premature withdrawals, laboratory parameters, and any other additional information as requested by the DSMB. Death or a life-threatening event in any study patient that was deemed by either the investigator or the sponsor to be at least possibly related to the study medication would trigger an evaluation by the DSMB for recommending stopping the study. Further details on the DSMB, schedule of interim assessments, and the Charter and members is provided in the Supplemental Methods.
Patients
Patients provided written informed consent before entering either study. Full eligibility criteria are provided in Supplemental Table 1. Enrolled patients were male or female, between 18 and 80 years of age, and diagnosed with early-stage PD within the previous 3 years. Early-stage PD was defined as the absence of motor fluctuations or L-dopa–induced dyskinesia. For SKY, patients had a body weight between ≥60 kg and ≤100 kg. Dopaminergic-treated patients in EMBARK and all patients in SKY were on a stable dose of a dopaminergic agonist MAO-B inhibitor or L-dopa, either alone or in combination up to the L-dopa equivalent dose of 600 mg daily for at least 3 months before screening (calculated according to the formula outlined in Tomlinson et al. 18 ). Key exclusion criteria included atypical or secondary parkinsonism without dopa-responsiveness, progressing (ie, disease that is unstable and is worsening) Axis I psychiatric disorders based on a semi-structured interview in accordance with the Diagnostic and Statistical Manual of Mental Disorders V, or disorders associated with neutropenia or thrombocytopenia. In addition, the therapeutic conditions of patients in the EMBARK study had to remain unchanged for the duration of the study, in accordance with the inclusion criteria for both dopaminergic-naïve and dopaminergic-treated patients (Supplemental Table 1).
Rationale for dosing
Data from two previous studies suggested that 15 mg/kg BID is the dose most likely to provide the greatest benefit versus risk, but few patients enrolled in those studies.10,19 Body weight is likely to vary less than 4-fold for the vast majority of patients with PD, and it was estimated based on clinical experience that the average body weight of patients with PD is about 80 kg. Using four fixed doses of 300, 600, 900, and 1200 mg BID (i.e., for total daily dosages of 600, 1200, 1800, and 2400 mg) and an inclusion criterion of body weight between 60 and 100 kg should result in actual doses ranging from 3–20 mg/kg BID centered on doses of 4–15 mg/kg BID for patients using an average weight of 80 kg. Twice-daily dosing was based on preclinical studies, suggesting no differences in efficacy with once-, twice-, or thrice-daily dosing; twice-daily was chosen to optimize tolerance based on results from the FAIRPARK-II study that demonstrated safety of twice-daily dosing. 17 Patients received delayed-release dosing specifically formulated for PD and other neurodegenerative disorders.
All patients in EMBARK received deferiprone oral solution (80 mg/mL) at a dose of 15 mg/kg BID, at least 8 h apart, for a total daily dosage of 30 mg/kg for up to 9 months.
Randomization and masking (SKY only)
Randomization codes were generated and finalized by the sponsor; an IVRS/IWRS was used for randomization. Randomization codes were not modifiable after generation but could be broken in the event of emergency, with code-breaking instructions provided to study sites. Eligible patients were randomized to one of the four deferiprone (or placebo-matching) dosage cohorts using an interactive voice/web response system. Each cohort was further randomized at a 4:1 ratio to receive either their assigned deferiprone dose or matching dose of placebo tablets (Figure 1). Patients were randomized to dosage cohorts in parallel (i.e., ascending dose cohorts were not used). The assignment of patients to each treatment or placebo group was blinded to patients, investigators, and staff at the study site.
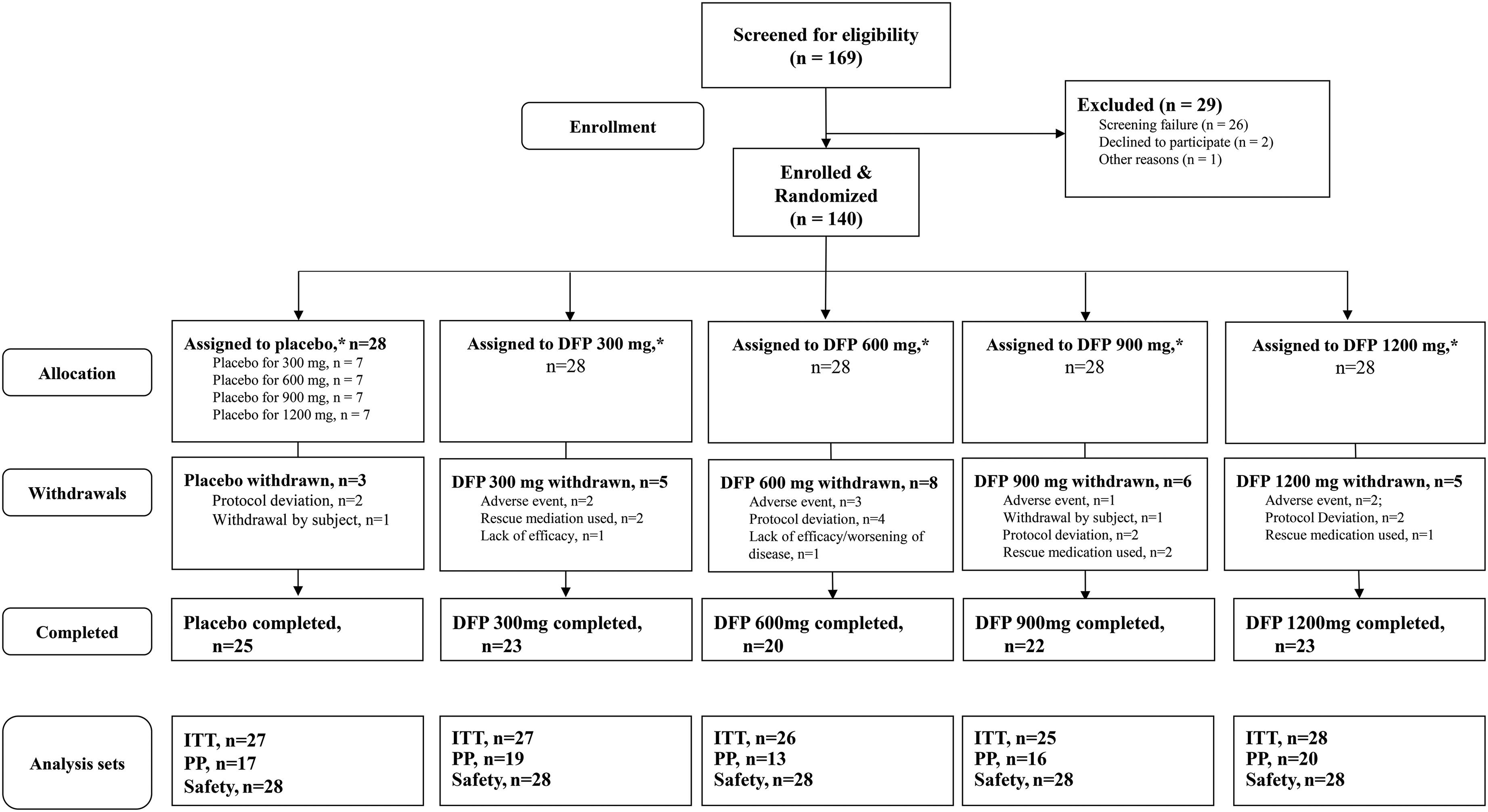
Patient disposition in the SKY study.
Procedures
The complete schedule of assessments for both studies can be found in Supplemental Table 2. Deferiprone delayed-release tablets or matching placebo tablets (SKY only) were administered orally to patients twice daily, at least 8 h apart, for 9 months. Efficacy was evaluated at baseline, and months 3, 6, and 9, using the MDS-UPDRS (Part III was evaluated in the “ON” condition as patients were receiving usual dopaminergic treatment and had a good response) and Montreal Cognitive Assessment (MoCA; SKY only). 20
Monitoring of all AEs occurred after the start of dosing throughout the study up to the safety follow-up at month 10 or on withdrawal. Safety was assessed through monitoring laboratory measures (hematology, blood chemistry, and urinalysis), electrocardiograms, vital signs, physical examinations, and suicidality throughout the study. Drop-out related to disease worsening was defined as patient-reported worsening of specific indications of PD such as bradykinesia, rigidity, tremor, gait, or a global disease worsening. Poststudy safety was assessed by a follow-up telephone call 1 month after the end of the study.
Outcomes
The primary efficacy outcome for both studies was the change from baseline to month 9 of the MDS-UPDRS motor examination subscore (Part III). Secondary efficacy outcomes for both studies included the change from baseline to month 9 in the MDS-UPDRS total score and subscale scores for nonmotor experiences of daily living (Part I), motor experiences of daily living (Part II), motor complications (Part IV), combined motor examinations (Parts II + III), 21 time to rescue medication, and safety.
Statistical analyses
For SKY, no formal sample size and power calculations were conducted; however, based on the results of FAIRPARK-I that demonstrated a significant change of the mean UPDRS Part III (
For EMBARK, the planned enrollment was 40 patients, with half dopaminergic-treated patients and half who were treatment naïve. Patients who withdrew before month 6 could be replaced. The treatment groups were analyzed as two separate subgroups. The ITT, PP, and safety populations were defined as in SKY; the ITT population was used for all efficacy analyses; the PP population was used only for the primary endpoint analysis.
For both studies, the change from baseline to each follow-up visit was summarized using descriptive statistics for each continuous efficacy endpoint. Frequency and proportions are presented for each discrete efficacy endpoint.
A Mixed-Effect Model Repeated Measure (MMRM) analysis assessed the effect of deferiprone on change from baseline to study end for all efficacy variables except time elapsed until rescue medication, which was presented using Kaplan-Meier survival curve and compared among treatment groups using the log-rank test. The MMRM model used baseline values as a covariate and treatment group and visit as the main factors. The MIXED procedure in SAS was used for the MMRM model analysis, and data from each patient at different visits were considered repeated measures. A first-order autoregressive covariance structure was used to model the correlation between repeated measures within the same patients, and the Kenward-Roger method was used to estimate the denominator degrees of freedom. The Least Square Mean (LSM) and 95% confidence interval (CI) of LSM difference was calculated by LSMEANS. For SKY, a regression analysis was used to assess the dose-response relationship.
For all efficacy measures except time elapsed until rescue medication, missing data were assumed to be missing at random (MAR). The MMRM model analyses were based on observed cases. A sensitivity analysis was conducted for the primary efficacy endpoint, with missing data being imputed as follows. In cases where early termination was due to either worsening of disease conditions or inadequate efficacy of the drug (as documented in the case report form), the “worst score” method was used to fill in missing data. With this method, the average change score for the placebo group at a particular visit was used to impute any missing data at that visit, while for the active group, the worst change score of all patients at that visit was used. In cases where early termination was due to some other reason, such as a missed visit, the last observation carried forward (LOCF) method was used to fill in the missing data. For the MDS-UPDRS and MoCA scores, missing data were calculated as outlined in Supplemental Methods.
SKY was registered with ClinicalTrials.gov, number NCT02728843, and the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT), number 2015-004344-19. EMBARK was registered with ANZCTR, number ACTRN12617001578392.
Role of the funding source
The sponsor of the studies was involved in study design, data collection, data analysis, data interpretation, and writing of the report.
Results
Baseline demographics (SKY)
Between July 22, 2016, and September 4, 2019, 140 patients with early-stage PD were enrolled and randomized to five dose cohorts: placebo, or twice-daily deferiprone 300 mg, 600 mg, 900 mg, or 1200 mg (n = 28 each; Figure 1). Overall, the mean ± SD age was 61.4 ± 9.0 years; 67.7% were men. There were no significant differences between groups in baseline demographics, MDS-UPDRS total or subscale scores, or MoCA cognitive assessment scores (Table 1).
Patient demographics and clinical characteristics at baseline (ITT population).
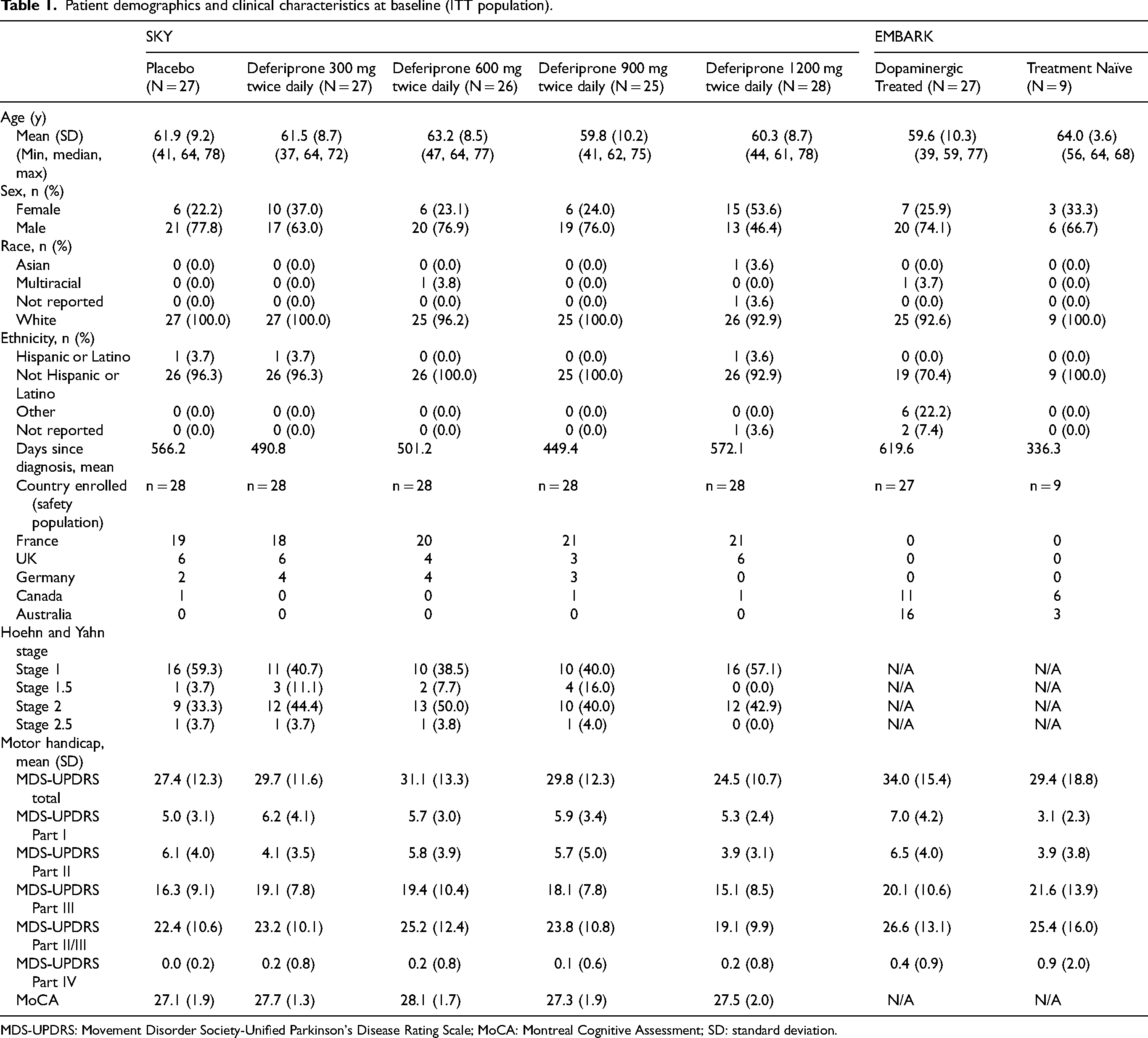
MDS-UPDRS: Movement Disorder Society-Unified Parkinson's Disease Rating Scale; MoCA: Montreal Cognitive Assessment; SD: standard deviation.
Patient disposition is shown in Figure 1. Study completion rates ranged from 71.4% (20/28) in the 600 mg group to 89.3% (25/28) in the placebo. Twenty-seven patients discontinued from the study; two withdrew themselves. The remainder were withdrawn due to protocol violations (n = 10; all of which involved inclusion criteria violation regarding their stable use of medication [i.e., discovery after randomization and dosing that patients were not taking antiparkinsonian drug at time of study entry] or changes in dosage or type of antiparkinsonian medication during the trial), AEs (n = 8), or worsening of PD symptoms (n = 7). Of the AEs that led to withdrawal, 5 were considered possibly related to study treatment: neutropenia (600 mg and 1200 mg groups), asthenia (300 mg group), and nausea (300 mg group). One case of asthenia was considered definitely related (600 mg group).
Baseline demographics (EMBARK)
The trial was terminated before reaching the planned enrollment of 40 patients due to difficulties in both enrollment and retention. Between November 28, 2016, and October 9, 2019, 36 patients enrolled: 27 into the dopaminergic-treated group (over-enrollment due to 7 patients who replaced patients who withdrew before Month 6), and 9 into the treatment-naïve group (none of whom were replacements). All 36 patients are included in the ITT and safety populations. The average age was 60.7 years; 72.2% were men (Table 1).
Patient disposition is shown in Figure 2. Sixteen (59.3%) patients in the dopaminergic-treated group and 3 (33.3%) in the treatment-naïve group completed the study. Eight patients withdrew from the study owing to AEs (n = 8), worsening of the disease (n = 5), use of rescue medication (n = 1), protocol deviation (n = 1), and voluntary withdrawal (n = 1). Of the AEs that led to withdrawal, 6 were considered at least possibly related to study treatment; however, none required rescue medication. In the naïve group, these events were increased prolactin (definitely related) and anorgasmia (possibly related); in the dopaminergic-treated group, they were agranulocytosis (definitely related and considered serious), nausea (definitely related), reflux esophagitis (probably related), and dyskinesia (possibly related). Of note, the dopaminergic-treated patient with agranulocytosis had been stable and closely monitored before the onset of this event, simultaneous escherichia sepsis and pseudomonal sepsis, and hospitalization with brain abscesses. These AEs were considered severe, at least possibly related to deferiprone, and resolved. The patient was withdrawn due to the agranulocytosis event, and deferiprone was discontinued.
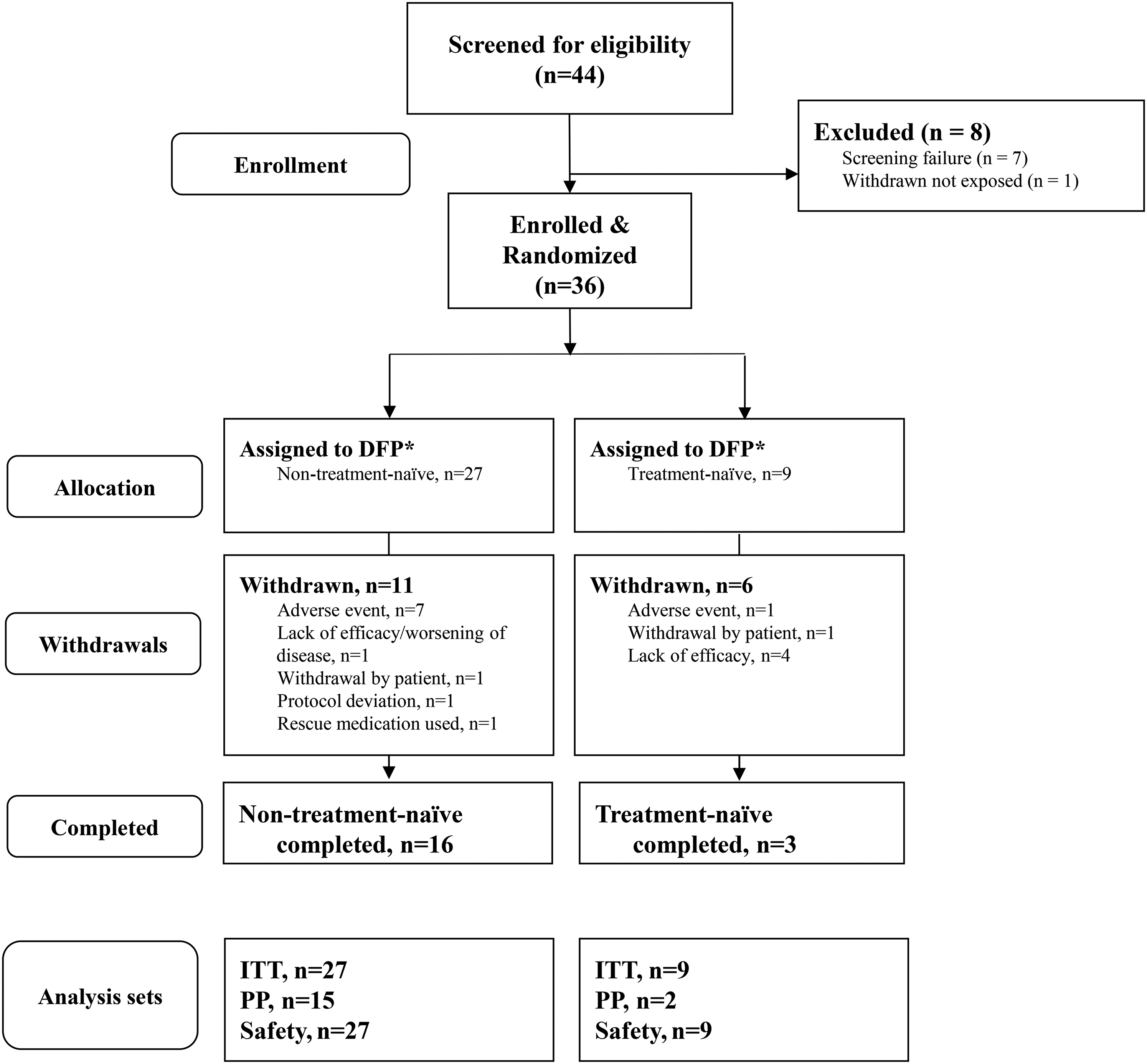
Patient disposition in the EMBARK study.
Efficacy (SKY)
For the primary endpoint of motor function (change from baseline to month 9 in MDS-UPDRS Part III score), all dosage groups, whether ITT or PP population, showed worsening over the 9-month treatment period, except for the 600 mg group with a nonsignificant trend toward improvement (Table 2). No dose-response effect was observed (Figure 3). Placebo-corrected LSM (95% CI) change from baseline to month 9 in MDS-UPDRS was 2.2 (–1.7, 6.0) for 300 mg, −2.8 (–6.8, 1.1) for 600 mg, 1.9 (–2.1, 5.8) for 900 mg, and 0.1 (–3.8, 4.0) for 1200 mg.
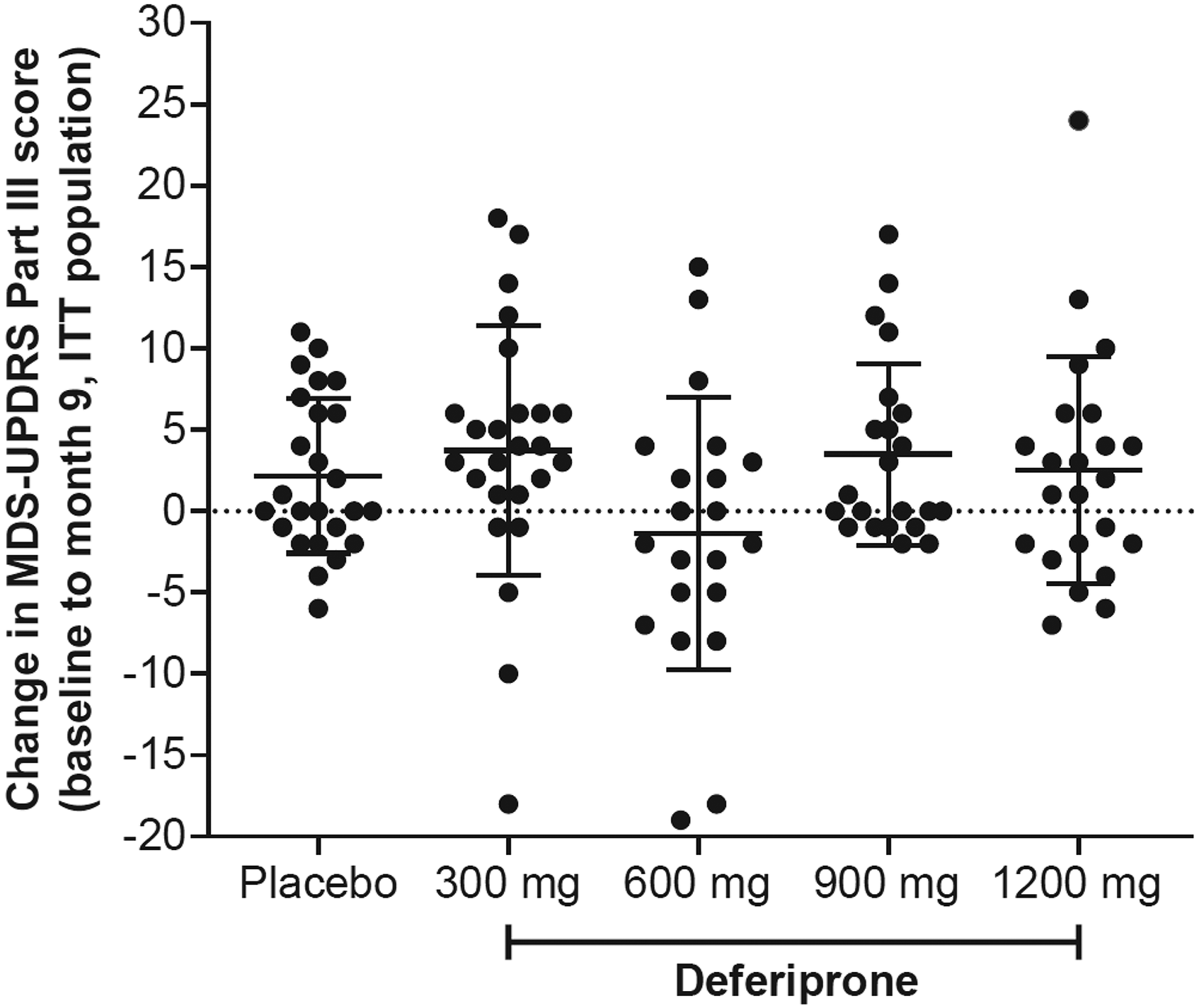
Change from baseline to month 9 in MDS-UPDRS Part III score by deferiprone dose (ITT population). Dots represent the change from baseline to month 9 in MDS-UPDRS Part III score for individual patients in each deferiprone dose group. Line represents group mean and error bars represent SD.
LSM change from baseline to month 9 in primary and secondary efficacy outcomes.
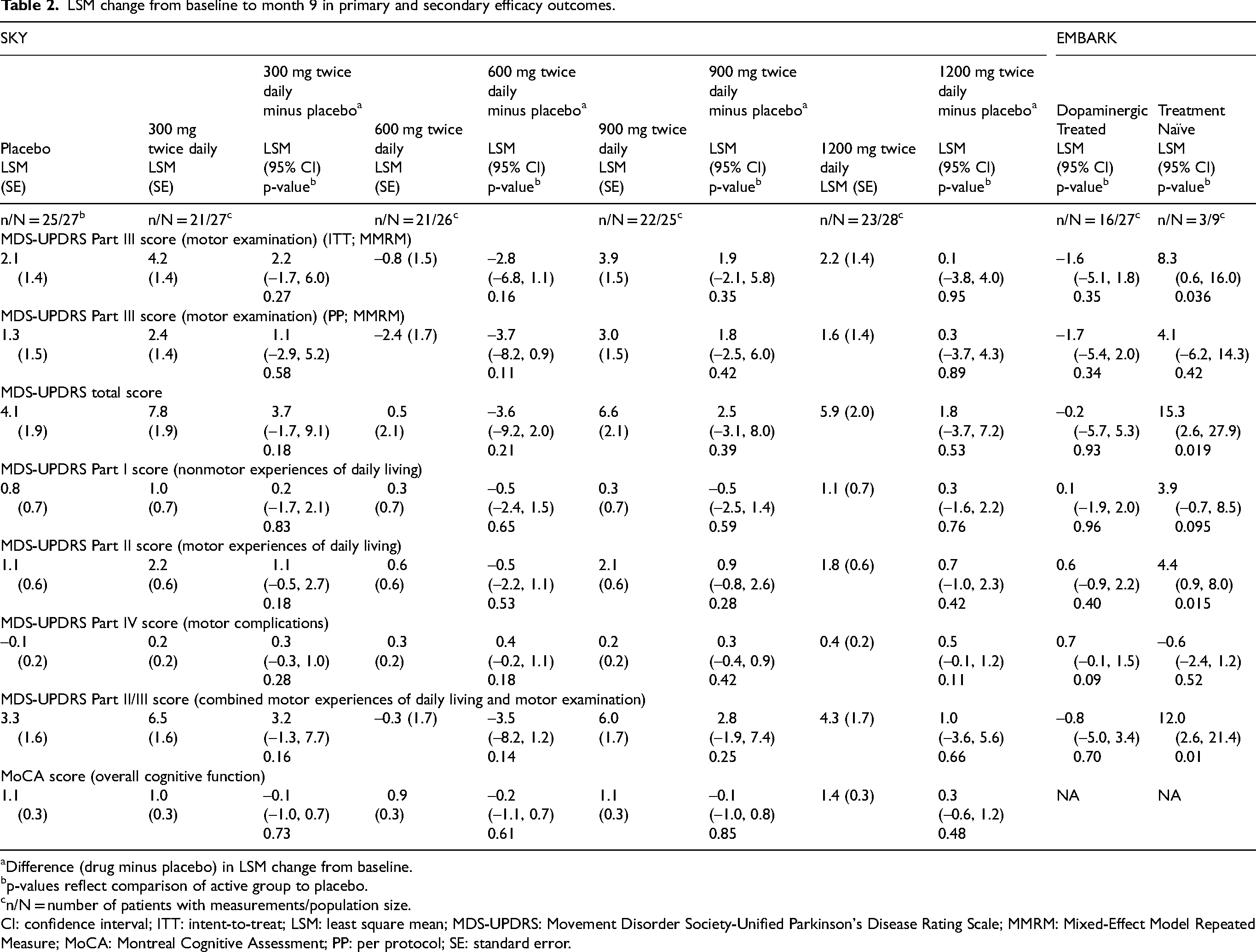
Difference (drug minus placebo) in LSM change from baseline.
p-values reflect comparison of active group to placebo.
n/N = number of patients with measurements/population size.
CI: confidence interval; ITT: intent-to-treat; LSM: least square mean; MDS-UPDRS: Movement Disorder Society-Unified Parkinson's Disease Rating Scale; MMRM: Mixed-Effect Model Repeated Measure; MoCA: Montreal Cognitive Assessment; PP: per protocol; SE: standard error.
The change from baseline to month 9 was similar between placebo and any of the deferiprone dose cohorts for MDS-UPDRS total score, Parts I, II, IV, or combined II + III scores (Table 2). No significant differences were observed in overall cognitive status (MoCA score) between the placebo and any of the deferiprone dose cohorts (Table 2). Five patients (2 in the 300 mg group, 2 in the 900 mg group, and 1 in the 1200 mg group) required rescue medication, and there were no group differences (p > 0.4).
Efficacy (EMBARK)
For the primary endpoint of LSM (SE) of change from baseline in MDS-UPDRS Part III, a nonsignificant decrease or improvement was observed at month 9 in the dopaminergic-treated group (−1.6 [1.7]), while statistically significant worsening was seen in the treatment-naïve group for the ITT population (8.3 [3.9]; Table 2). Similar nonsignificant trends were observed for the PP population.
Changes in secondary endpoints are shown in Table 2. In the dopaminergic-treated group, stable changes or worsening were seen for all measures except Part IV, while in the naïve group, worsening was seen for all measures except Part IV. Only one patient in the dopaminergic-treated group required rescue medication, and there was no group difference.
Additional secondary endpoints (SKY and EMBARK)
Results of additional secondary endpoints, including pharmacokinetics and pharmacodynamics analyses from both studies are shown in the Supplemental Material.
Post hoc analysis (SKY)
A post hoc analysis was conducted for SKY that recategorized patients according to their equivalent dose in mg/kg, which showed that mean change in MDS-UPDRS Part III score from baseline to month 9 was similar across the deferiprone and placebo groups, and safety was similar to previous studies (Supplemental Table 11).
Safety (SKY)
Mean percentage ± SD adherence was high across all groups at the final visit, ranging from 89.1% ± 23.6% to 95.8% ± 13.2%. Total exposure to deferiprone ranged from 17.2 to 19.3 person-years, with a mean duration of drug exposure ranging from 0.6 to 0.7 years.
The number of AEs reported in the placebo and deferiprone groups is listed in Table 3. Among the patients who received deferiprone, the percentage of those who reported at least one AE of any type ranged from 82.1% (n = 23, 600 mg) to 89.3% (n = 25, 1200 mg) compared with 92.9% (n = 26) in the placebo group. The most common AEs were upper abdominal pain, constipation, diarrhea, nausea, toothache, asthenia, aggravated condition, fatigue, arthralgia, back pain, pain in extremity, dizziness, headache, syncope, tremor, oropharyngeal pain, and hypertension (Supplemental Material). The 1200 mg deferiprone group generally reported more gastrointestinal-related AEs than the lower dose deferiprone groups and placebo. Eleven patients reported serious adverse events (SAEs) in the placebo and deferiprone groups, which are listed in Table 3. SAEs occurring in more than 1 patient were neutropenia (1 from each of the deferiprone 600 mg, 900 mg, and 1200 mg dose groups) and atrial fibrillation (1 each in the placebo and deferiprone 900 mg group; Supplemental Material).
Aes and SAEs – Serious and severe (Safety population).
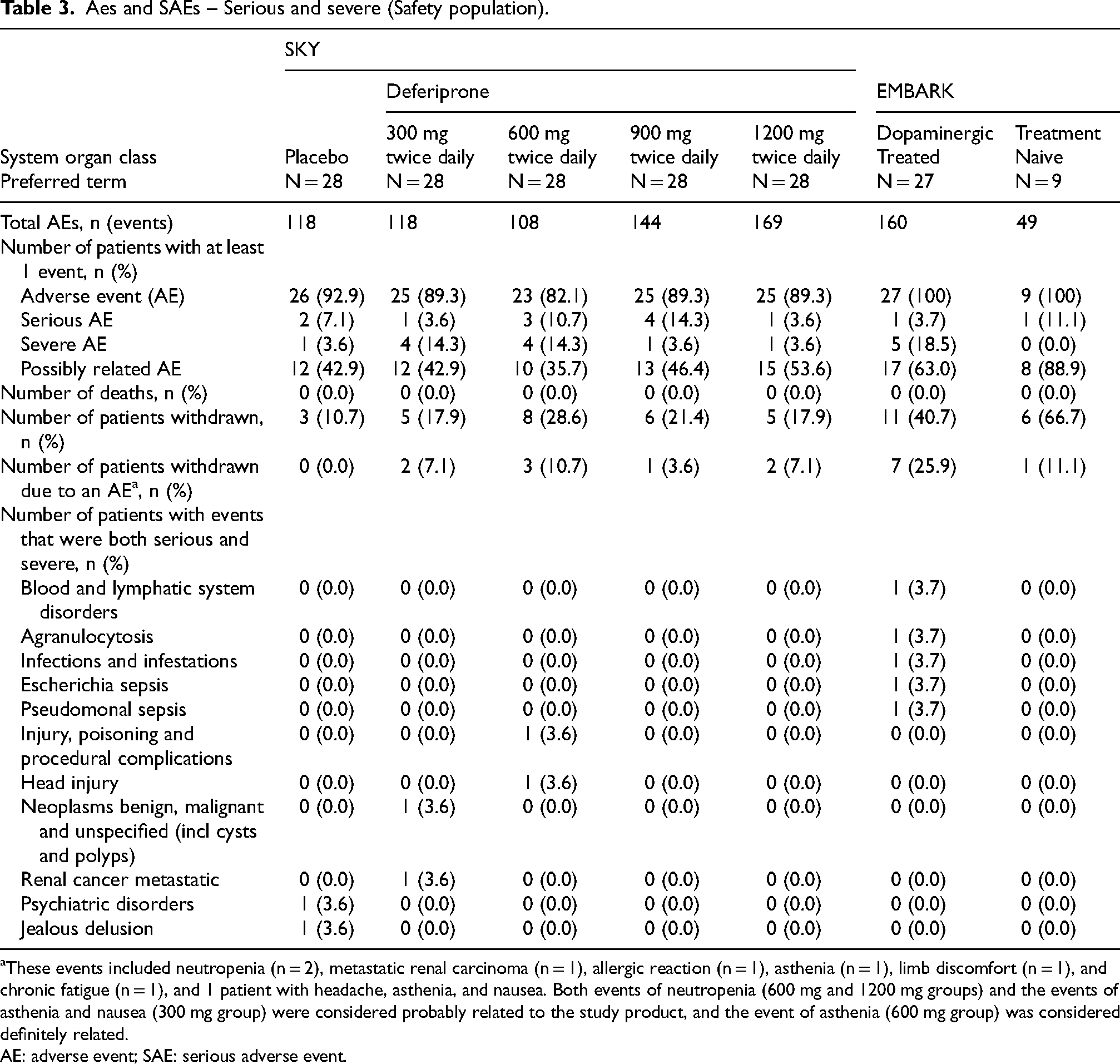
These events included neutropenia (n = 2), metastatic renal carcinoma (n = 1), allergic reaction (n = 1), asthenia (n = 1), limb discomfort (n = 1), and chronic fatigue (n = 1), and 1 patient with headache, asthenia, and nausea. Both events of neutropenia (600 mg and 1200 mg groups) and the events of asthenia and nausea (300 mg group) were considered probably related to the study product, and the event of asthenia (600 mg group) was considered definitely related.
AE: adverse event; SAE: serious adverse event.
AEs considered at least possibly related to deferiprone were identified as adverse drug reactions (ADRs) and are listed in Supplemental Table 4. Most ADRs did not occur in more than one patient in any treatment group. Those observed in more than one patient per group included lymphopenia and hypertension in the placebo group; aggravated PD, asthenia, and tremor in the deferiprone 300 mg group; nausea, headache, asthenia, and decreased neutrophil count in the 600 mg group; asthenia, pain in extremity, headache, and tremor in the 900 mg group; and diarrhea, fatigue, lymphopenia, constipation, nausea, decreased neutrophil count, and headache in the 1200 mg group.
Safety (EMBARK)
Adherence was good overall: for the dopaminergic-treated group, mean adherence ranged from 97.9% to 99.0% up to month 6 and was 91.3% at the final visit. In the naïve group, the rates were 91.1% to 95.3% and 83.0%, respectively. Total exposure to deferiprone ranged from 0.01 to 0.76 person-years, with mean exposure of 0.57 years in the dopaminergic-treated group and 0.40 years in the treatment-naïve group.
AEs reported in EMBARK are shown in Table 3 and Supplemental Table 3. All patients experienced at least one AE of any type; the most common were worsening disease (33.3% each group), headache (25.9% dopaminergic-treated and 33.3% in treatment-naïve), and nausea (22.2% dopaminergic-treated). One patient in each group had a serious AE; 5 (18.5%) dopaminergic-treated patients vs. no treatment-naïve patients experienced an AE of severe intensity; and 17 (63.0%) vs. 8 (88.9%) had an AE at least possibly related to deferiprone. In the dopaminergic-treated group, the only ADRs seen in more than 1 patient were neutrophil count decreased, weight increased, and headache (3 patients each), and nausea, fatigue, alanine aminotransferase increased, and aspartate aminotransferase increased (2 patients each). In the treatment-naïve group, the only ADRs seen in more than 1 patient were blood prolactin increased (3 patients) and fatigue (2 patients). ADRs occurring in at least 1 patient in the EMBARK study are listed in Supplemental Table 4. Seven (25.9%) dopaminergic-treated patients vs. 1 (11.1%) treatment-naïve patient were withdrawn from the study because of an AE. There were no deaths.
Discussion
After pilot studies in patients with early-stage PD on stable dopaminergic medication suggesting benefit of deferiprone,10,16 and a significant worsening of disease in dopaminergic treatment-naïve patients, 17 there was a need for additional studies to examine the potential of deferiprone to reduce PD progression in a dopaminergic-treated patient population. The SKY study found that 600 mg twice-daily deferiprone was the only dose associated with a nonsignificant reduction of MDS-UPDRS Part III score from the baseline compared with placebo. The EMBARK study was designed to provide additional information on the effect of deferiprone in dopaminergic-treated versus treatment-naïve patients and showed a nonsignificant trend toward improvement in MDS-UPDRS Part III score from the baseline in patients on stable dopaminergic therapy, and a significant worsening of disease in treatment-naïve patients. Overall, these results support previous findings that deferiprone administered to patients naive to dopaminergic treatment were more likely to observe significant worsening; 17 however, when administered to patients with ongoing dopaminergic treatment, no worsening of disease was observed.6,10 As suggested in the FAIRPARK-II study, 17 a possible mechanistic scenario to explain these findings is that exogenous dopaminergic treatment could be compensating for a presumed decrease in endogenous dopamine resulting from deferiprone treatment and as indicated by increased prolactin levels.
Two previous pilot monocentric studies showed a positive symptomatic effect with a liquid formulation of deferiprone in patients with early-stage PD also receiving dopaminergic treatments.10,16 One study showed a significant 3-point improvement compared with placebo in UPDRS Part III score after 6 months with the deferiprone dose of 15 mg/kg BID. 10 In contrast, the second study showed a nonsignificant trend toward improvement of MDS-UPDRS Part III score with doses of 10 and 15 mg/kg BID. 16 In the SKY study, the twice-daily deferiprone fixed dose of 600 mg (corresponding to a liquid dose of ∼6 to 10 mg/kg BID) showed a nonsignificant trend toward improvement with a good safety profile. Indeed, the upper doses of 900 mg and 1200 mg BID (equivalent on average to doses of 11.5 and 15 mg/kg BID) and lower dose of 300 mg BID (below 5 mg/kg BID) showed no difference or worsening compared to placebo. Deferiprone treatment of another neurodegenerative disorder, Friedreich ataxia, found that doses between 10 and 20 mg/kg BID could effectively decrease iron accumulation in the dentate nuclei. 14 In EMBARK, patients received 15 mg/kg BID. Findings from the SKY study indicate a lower dose (600 mg BID [i.e., ∼6 to 10 mg/kg BID]), which showed nonsignificant improvement, could be chosen in future trials to assess the potential of deferiprone to reduce the progression of PD.
In EMBARK and SKY, deferiprone had a safety profile in patients with early-stage PD that was similar to previous reports in patients with thalassemia and sickle-cell disease.22–24 Most AEs observed in the SKY and EMBARK were nonspecific, gastrointestinal, or disease-related. Gastrointestinal AEs, such as nausea, vomiting, and abdominal pain/discomfort, are consistently reported with iron chelators.25,26 In SKY, gastrointestinal-related AEs were reported by more patients receiving the highest dose of deferiprone compared with the lower dose deferiprone groups and placebo. The prevalence of nervous system disorders is consistent with PD symptomology and was not different from placebo group in the SKY study, meaning no PD worsening. 27 The most SAEs associated with deferiprone are severe neutropenia or agranulocytosis. Across both studies, only one patient in the dopaminergic-treated group developed agranulocytosis, which led to study withdrawal and occurred along with sepsis and brain abscesses that required hospitalization and several months of antibiotic treatment. The rate of neutropenia in SKY was consistent with those in thalassemia populations receiving deferiprone,28,29 and all incidences of neutropenia that led to study withdrawal in SKY resolved with drug discontinuation. Only one case of anemia (in SKY; none were reported in EMBARK) was reported with the highest dose of deferiprone, which suggests conservative iron chelation. 27 Finally, rates of withdrawal from the studies ranged from 20% to 40% among patients on dopaminergic therapy, frequently owing to AEs or disease aggravation; however, for those who remained on therapy, adherence was fairly high, ranging from 80% to 90%. The need for rescue medication was also low in both studies.
The results of this study suggest complex relationships between neuronal iron homeostasis and the interaction between iron and dopamine in the context of PD. The lack of benefit with the lower and higher dose groups in SKY suggests a possible inverted U-shaped dose-response profile centered on the fixed dose of 600 mg BID; however, despite showing trends of improvement the results for 600 mg BID were nonsignificant. It is possible that iron levels in patients with PD may need to be maintained within a certain range. Iron is an important component of tyrosine hydroxylase-dependent dopamine synthesis and other reactions associated with dopamine metabolism, so a certain level of iron is required for dopamine synthesis (and other functions). 4 However, several lines of evidence suggest that iron levels above a certain limit can cause neurotoxicity in PD. 4 Further research is needed to better understand this balance in patients with PD.
The results from EMBARK and previous studies demonstrate the importance of the interaction between dopamine and iron levels in the nigrostriatal pathway. Recent results from the FAIRPARK-II study demonstrated that 15 mg/kg BID of deferiprone in dopaminergic therapy-naive patients worsened their MDS-UPDRS Part III motor score by 5.8 points compared with placebo over 9 months, consistent with the results in the treatment-naive patients from EMBARK. 17 The lack of placebo arm to compare the deterioration of the treatment-naive group against, as well as insufficient and unbalanced data between dopaminergic-treated patients (ITT = 27, PP = 16) and the treatment-naive group (ITT = 9, PP = 3), means that caution is required to interpret the results. As suggested in the FAIRPARK-II study, the increase in prolactin levels, a marker of dopamine inhibition, with deferiprone treatment may indicate decreased dopamine synthesis; this was speculated to be due to iron chelation reducing activity of tyrosine hydroxylase, the rate-limiting enzyme for dopamine production. 17 In EMBARK, nonsignificant increases in prolactin were observed 2 h postdose in the dopaminergic-treated group (p = 0.2136), whereas significant increases in prolactin were observed 2 h postdose in the treatment-naïve group (p = 0.0004). The association of significantly increased prolactin levels with deferiprone in treatment-naive patients, but not dopaminergic-treated patients, provides further support to the mechanistic hypothesis outlined in the FAIRPARK-II study and suggests that excess iron in the nigrostriatal pathway of patients with PD may be an adaptive mechanism to increase the production of dopamine. 30 However, further studies are required to confirm this hypothesis.
The main limitation of SKY was that, despite the large number of patients (140) enrolled, the evaluation of four deferiprone doses reduced the statistical power with 13 to 27 patients per group. Furthermore, drop-out rates ranged from 20% to 40% and differed across groups, which may have influenced results. However, of the 28 patients treated with the fixed dose of 600 mg BID, only 1 patient (3.6%) withdrew due to PD progression, and 3 patients (11%) withdrew for an AE unrelated with PD progression with the fixed dose of 600 mg BID. The small number of patients per treatment cohort limits confidence that the 600 mg is the correct dose; this dosage is also below to within range of beneficial doses seen in EMBARK and previous studies. Future studies may explore a personalized medicine approach to calibrate according to body weight or baseline brain iron levels. Another limitation was that participants were aware of which dosing cohort they were in because of the different number of tablets assigned; however, within that cohort they did not know whether they were receiving active product or placebo. In addition, the slow progression of PD over a limited period of 9 months (leading to only 2.1 points of MDS-UPDRS Part III progression in the placebo group) may have contributed to a reduced likelihood of observing a significant change in disease progression with deferiprone treatment. In EMBARK, study limitations include its termination before planned enrollment was reached, with only 9 treatment-naïve patients successfully enrolled. Furthermore, EMBARK was not a blinded, randomized, study but was open label. Both studies included patients who were predominantly White and in the first 3 years of illness, which does not allow the conclusions to be extended beyond this population. Other limitations include lack of magnetic resonance imaging to confirm reduction of iron levels, which limits interpretation of the results.
In conclusion, evidence from the SKY and EMBARK studies, together with evidence from previous studies, suggest that the excess iron in the nigrostriatal pathway of patients with PD may be an adaptive mechanism for increased production of dopamine. Administration of deferiprone 600 mg and 15 mg/kg twice daily for up to 9 months in the SKY and EMBARK studies, respectively, did not significantly worsen MDS-UPDRS scores from baseline in dopaminergic-treated patients. The EMBARK study confirms a worsening of disease in treatment-naive patients. Furthermore, no new safety concerns were raised by either study. In the event of future studies evaluating the ability of deferiprone or other iron chelator to reduce the progression of PD, it would be advisable to consider 1) a larger population of patients with early-stage PD, 2) that they receive dopaminergic treatment 3) to choose a low dose of deferiprone (e.g., 600 mg twice a day) or other iron chelator and 4) to evaluate over the longer term.
Supplemental Material
sj-pdf-1-pkn-10.1177_1877718X241300295 - Supplemental material for Therapeutic modalities of deferiprone in Parkinson's disease: SKY and EMBARK studies
Supplemental material, sj-pdf-1-pkn-10.1177_1877718X241300295 for Therapeutic modalities of deferiprone in Parkinson's disease: SKY and EMBARK studies by David Devos, Olivier Rascol, Wassilios G Meissner, Alexandra Foubert-Samier, Simon Lewis, Christine Tranchant, Mathieu Anheim, David Maltête, Philippe Remy, Karla Eggert, Heidi Pape, Christian Geny, Philippe Couratier, Camille Carroll, Ray Sheridan, David Burn, Nicola Pavese, Jason Raw, Daniela Berg, Oksana Suchowersky, Lorraine V Kalia, Andrew Evans, Sophie Drapier, Teodor Danaila, Alfons Schnitzler, Jean-Christophe Corvol, Gilles Defer, Noemi Toiber Temin, Caroline Fradette, Fernando Tricta, Caroline Moreau and in Journal of Parkinson's Disease
Supplemental Material
sj-docx-2-pkn-10.1177_1877718X241300295 - Supplemental material for Therapeutic modalities of deferiprone in Parkinson's disease: SKY and EMBARK studies
Supplemental material, sj-docx-2-pkn-10.1177_1877718X241300295 for Therapeutic modalities of deferiprone in Parkinson's disease: SKY and EMBARK studies by David Devos, Olivier Rascol, Wassilios G Meissner, Alexandra Foubert-Samier, Simon Lewis, Christine Tranchant, Mathieu Anheim, David Maltête, Philippe Remy, Karla Eggert, Heidi Pape, Christian Geny, Philippe Couratier, Camille Carroll, Ray Sheridan, David Burn, Nicola Pavese, Jason Raw, Daniela Berg, Oksana Suchowersky, Lorraine V Kalia, Andrew Evans, Sophie Drapier, Teodor Danaila, Alfons Schnitzler, Jean-Christophe Corvol, Gilles Defer, Noemi Toiber Temin, Caroline Fradette, Fernando Tricta, Caroline Moreau and in Journal of Parkinson's Disease
Footnotes
Acknowledgments
ApoPharma Inc. (now Chiesi, Inc) designed and conducted the trial. The Chiesi Protocol ID: LA48-0215 and LA52-0215; Clinical trial: NCT02728843 and ANZCTR, number ACTRN12617001578392. The French NS-Park network assisted in study recruitment in France. The authors wish to thank the members of the DSMB: Prof Giovanni Abbruzzese, Dr Alan Cohen, Dr Gian Luca Forni, and Dr Robert Hauser. Medical writing support was provided by Marisa DeGuzman, PhD, Ebony Lai Hing, PhD, Leanne M. Low, PhD, and Michael Dyle, PhD, of Oxford PharmaGenesis Inc., Newtown, PA, USA, and funded by Chiesi USA, Inc.
ORCID iDs
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Chiesi Canada Corp. (formerly ApoPharma Inc.).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
The remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
All data requests should be submitted to the corresponding author for consideration. Chiesi will approve or deny data requests from external parties on a case-by-case basis. Chiesi reserves the right to deny requests for any and all legally appropriate reasons. Data requests that risk sharing participant-level data or proprietary information will not be approved.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.