
Abstract
Background
In pediatric age, the PRKN mutation is reported as one of the most common genetic causes of Parkinson's disease. However, detailed clinical data on PRKN patients with pediatric onset are scarce.
Objective
To describe clinical characteristics, disease progression, and management of PRKN patients with pediatric onset.
Methods
PRKN patients with onset of clinical signs before the age of 18 years were included in this retrospective multicenter study. Collected data included detailed clinical characteristics, progression, and disease management. Data presentation is descriptive due to the sample size.
Results
Nine patients (five females) were included from five French movement disorders centers. The mean age at symptom onset was 10.78 ± 2.22 years (median, 11; range, 7–14). Dystonia was the first most common motor symptom (six patients). The mean time from symptom onset to genetic diagnosis was 13.33 ± 9.21 years (median, 11; range, 3–32). The most commonly reported non-motor symptoms were sleep disorders (seven patients), anxiety (six patients), and depression (five patients). The first treatment was L-dopa in four patients, dopamine agonist in two, carbamazepine in two, and rasagiline in one. Dyskinesia and impulse control disorders were the most common treatment-related side effects (nine and six patients, respectively). Four patients underwent deep brain stimulation surgery. The last available follow-up was at 27.22 ± 14.05 years (median, 28; range, 6–56) after the diagnosis.
Conclusions
This is the first study reporting detailed clinical features and long-term management of PRKN patients with pediatric onset. Prompt diagnosis and appropriate treatment strategies are important to optimize disease management.
Plain language summary
PRKN gene mutation is one of the most common genetic causes of Parkinson's disease (PD) in young age. However, there is a lack of detailed information about the characteristics of this mutation in children. The objective was to describe the clinical aspects, progression, management, and therapy in people who started with this type of PD in their childhood. The study was conducted in five French hospitals, and included patients who showed signs of the disease before the age of 18 years. Available information on disease presentation, progression, and management was collected and analyzed. Nine patients (five females) were included in the study. The average age at which symptoms started was at around 11 years. An abnormal posture (dystonia) of one leg was the most common first symptom. There was a delay of about 13 years from the onset of the symptoms to the final diagnosis. The most common non-motor symptoms were sleep disorders, followed by anxiety, and depression. The first treatment was L-dopa in four children, dopamine agonists in two, carbamazepine in two, and rasagiline in one. All children experienced side effects from the treatment, such as uncontrolled movements (dyskinesia), and behavioral issues (impulse control disorders). Four patients underwent deep brain stimulation surgery to help manage their symptoms. Patients were followed for about 28 years since the initial diagnosis. This is the first study providing detailed clinical features of people with PRKN mutation and pediatric onset symptoms, and their long-term follow-up. Prompt diagnosis and appropriate treatment strategies are important to optimize disease management.
Introduction
Parkinson's disease (PD) predominantly affects the elderly population, with a prevalence that significantly increases with age, reaching around 1% in individuals over the age of 60 years. 1 Major clinical PD features include motor symptoms such as resting tremor, bradykinesia, rigidity, and non-motor symptoms such as mood and sleep disorders, cognitive impairment, and dysautonomia. 2 Although the diagnosis is commonly made when motor symptoms appear, non-motor symptoms often occur before the onset of motor symptoms, and contribute to the disability caused by the disease. 3
Age is one of the most important risk factors for PD, with a mean age of motor symptoms’ onset in the mid-60 s. 4 Indeed, PD is rare in the young population, with only 3–7% of PD cases starting before the age of 50 years (defined as “early-onset” PD). 4 Overall, early-onset PD patients have a less aggressive disease progression and a longer disease course.4–9 Furthermore, early-onset PD has been subdivided into “young-onset” PD, which occurs between 21 and 50 years of age, and “juvenile PD’’, which begins before the age of 21 years.4–6 Some clinical and genetic differences can support this classification. Young-onset PD is less likely to be hereditary compared to juvenile PD, and its clinical features are usually similar to idiopathic PD.6–8 In contrast, juvenile PD is rarer, often linked to genetic mutations, and may present with atypical features.5,6,9 Several genes have been linked to juvenile PD, the most frequent being PRKN, PINK1, and DJ1. 10 These monogenic forms exhibit clinical heterogeneity and overlapping atypical features, which can delay the diagnosis. 11 Indeed, the frequent initial presentation with lower limb dystonia and gait impairment can lead to the misdiagnosis of L-dopa responsive dystonia or paroxysmal exercise-induced dystonia.6,11
PRKN-related PD is associated with an autosomal recessive inheritance pattern. 12 The PRKN gene encodes for the Parkin protein, a proteasomal E3 ubiquitin ligase, which regulates a variety of cellular processes such as neuronal apoptosis, mitochondrial homeostasis, and mitophagy. 13 PRKN mutation carriers are known to have lower limb dystonia, gait impairment, and early falls as initial symptoms, along with a slower PD progression.5,7,9 Motor fluctuations and dyskinesia appear rapidly after starting treatment with L-dopa, even under low doses. 11 However, PRKN-related PD exhibits a diverse spectrum of phenotypical variability in both presentation and disease progression. 14
The incidence of PRKN-related PD tends to rise with a decreasing age of onset. In different age groups, the frequency of PRKN-related PD was reported as 35–77% in the 0–20 years group, 17–26% in the 21–30 years group, and 2–3% in the 31–40 years one.5,9,15 However, especially in the pediatric population (generally defined between 0 and 18 years of age), initial misdiagnosis is common, and diagnosis can be delayed until adulthood, with an average delay of 25.30 ± 17.00 years.16,17 Therefore, PRKN-related PD with pediatric onset (poPRKN) and its phenotype need to be better recognized by pediatric neurologists.
Our study aims to describe the clinical characteristics, progression over the years, and overall management of this unique population.
Methods
This retrospective multicenter study was coordinated by the Grenoble Alpes University Hospital. Data were collected from the hospital medical records of five French movement disorders centers (Besancon, Grenoble, Lille, Limoges, and Paris) between January 2004 to June 2022. The study protocol was approved by the center for clinical research and innovation (Grenoble Alpes University Hospital) with reference number 38RC22.0034. Informed consent was obtained from all subjects.
Subjects
Patients with a confirmed PRKN gene mutation and a disease onset before the age of 18 years were included in the study.
Collected clinical data included age at symptoms’ onset, type of initial symptoms, age at diagnosis, disease duration up to the last follow-up, motor and non-motor symptoms, scores of the Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS-UPRDS) part III, the Montreal Cognitive Assessment MoCA), the Mini-Mental State Examination (MMSE), treatment (medical and surgical), and treatment side effects. Results of brain magnetic resonance imaging (MRI), dopamine transporter (DAT) scan, and genetic tests were also collected.
Detailed description of each subject is provided in the Supplemental Material.
Statistical analysis
Data presentation and analysis are descriptive due to the small sample size. Mean and standard deviation (SD), median and range were used to present numeric data.
Results
Nine patients (five females, four males) met the inclusion criteria. Clinical characteristics of the individual cases are summarized in Table 1.
Main clinical characteristics of poPRKN patients.
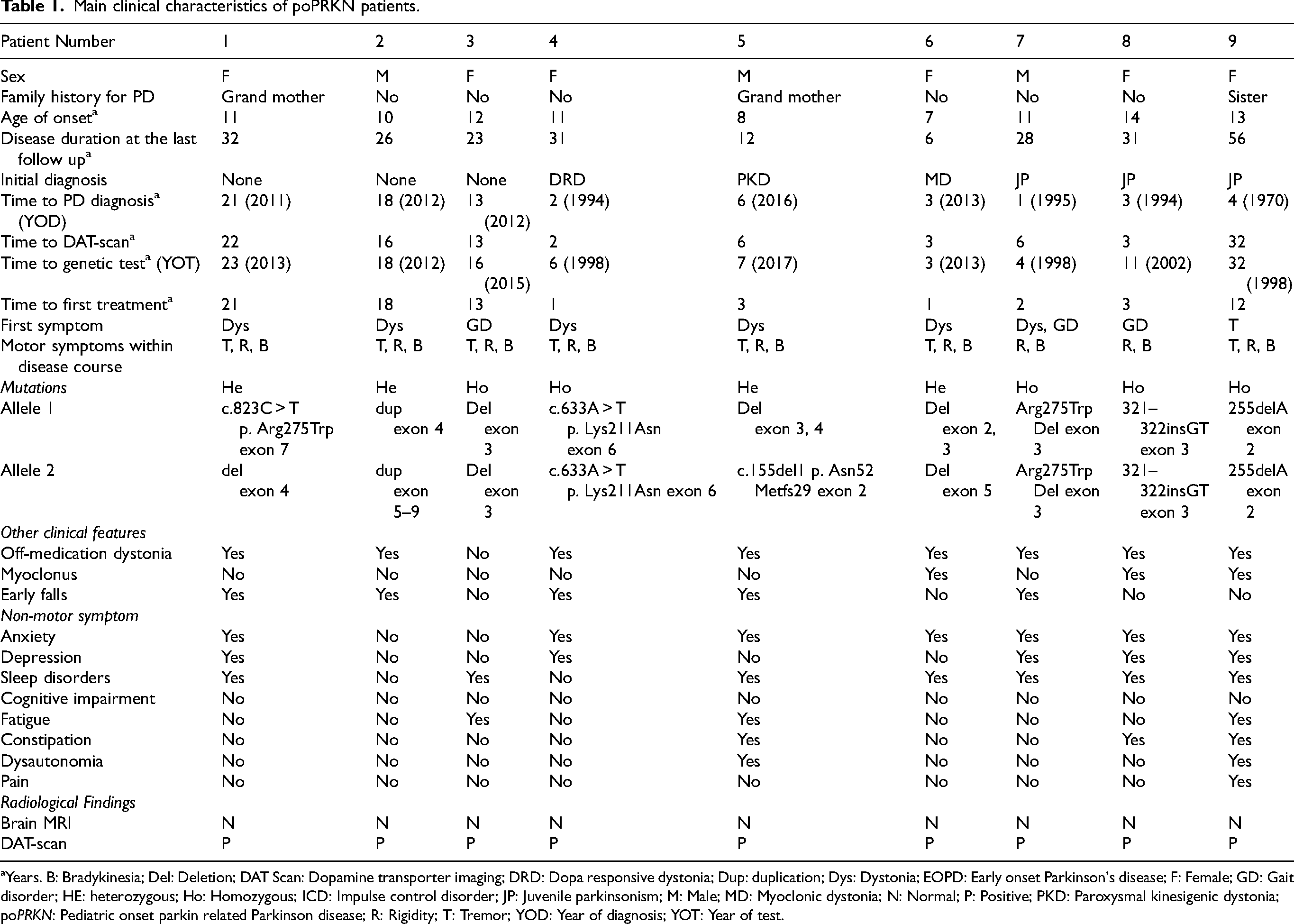
Years. B: Bradykinesia; Del: Deletion; DAT Scan: Dopamine transporter imaging; DRD: Dopa responsive dystonia; Dup: duplication; Dys: Dystonia; EOPD: Early onset Parkinson's disease; F: Female; GD: Gait disorder; HE: heterozygous; Ho: Homozygous; ICD: Impulse control disorder; JP: Juvenile parkinsonism; M: Male; MD: Myoclonic dystonia; N: Normal; P: Positive; PKD: Paroxysmal kinesigenic dystonia; poPRKN: Pediatric onset parkin related Parkinson disease; R: Rigidity; T: Tremor; YOD: Year of diagnosis; YOT: Year of test.
Three patients (33%) had a positive family history of PD. Two patients (patients 1 and 5) had their grandmother with the disease, and one patient (patient 9) had a sister with PD started at the age of 40 years (Table 1).
Mean age at symptoms’ onset was 10.78 ± 2.22 years (median, 11; range, 7–14).
Three patients (33%) were initially diagnosed as dystonia (myoclonic dystonia, dopa responsive dystonia, paroxysmal kinesigenic dystonia). In three patients (33%), there was no defined initial diagnosis until when they had an adult neurology consultation several years (13, 18, and 21 years) after the symptoms’ onset (Table 1). Three patients (33%) received the initial diagnosis of JPD. The mean time from the first symptom's onset to the genetic diagnosis was 13.33 ± 9.21 years (median, 11; range, 3–32). The year of the clinical diagnosis of PD, and the year of genetic testing for each patient are shown in Table 1.
The mean disease duration at the last available follow-up since the diagnosis was 27.22 ± 14.05 years (median, 28; range, 6–56 years).
Other clinical details of the long-term follow-up are specified in Table 2.
First and last available total scores of the MDS-UPDRS part III (ON-medication condition), and some information about common life activities of poPRKN patients at the last follow-up.

MDS-UPDRS III: Movement Disorders Society Unified Parkinson's disease rating scale part III; NA: not applicable.
Motor features
The most common first sign was foot dystonia (six patients, 66%) followed by gait impairment (three patients, 33%). Akinetic-rigid syndrome (two patients, 22%) and resting tremor (one patient, 11%) were relatively rare at disease onset. Early falls due to impaired balance were reported in five patients (55%) within the first five years of the disease. Seven patients (77%) developed resting tremor within the disease course. All patients presented with akinetic-rigid syndrome, off-dystonia, and troublesome peak-dose dyskinesia along the disease course. The first and the last available MDS-UPDRS part III scores are provided in Table 2. Other less common motor features are reported in Table 1.
Non-motor features
The average time until the onset of the first non-motor symptom was 13.00 ± 9.05 years (median, 13; range, 1–31 years). Sleep disorders (insomnia in five patients, fragmented sleep in three patients, REM sleep behavior disorder in one patient, and excessive daytime sleepiness in one patient) were the most common non-motor symptoms (seven patients, 77%), followed by anxiety (six patients, 66%), and depression (five patients, 55%). Fatigue was noted in three patients (33%), and constipation in three patients (33%). Dysautonomia was rarely reported (two patients, 22%). Other less common non-motor features are reported in Table 1.
Genetic data
Homozygous pathogenic variants were identified in five patients, while the remaining four cases had compound heterozygous variants falling in distinct exons of the gene. Details about genetic test results for each patient are shown in Table 1.
Radiological findings
All patients had normal brain MRI. The DAT-scan revealed symmetric presynaptic dopaminergic deficits in all patients. The mean time from the first symptom to the DAT-scan was 11.44 ± 9.98 years (median, 6; range, 2–32) (Table 1).
Treatment and related complications
The mean age at first treatment was 18.78 ± 8.73 years (median, 17; range, 8–32). First and last dopaminergic treatment regimens, surgical treatments, and treatment-related side effects are summarized in Table 3.
First and last dopaminergic treatment, treatment response, main treatment-related side effects, and device aided therapies of the poPRKN patients.
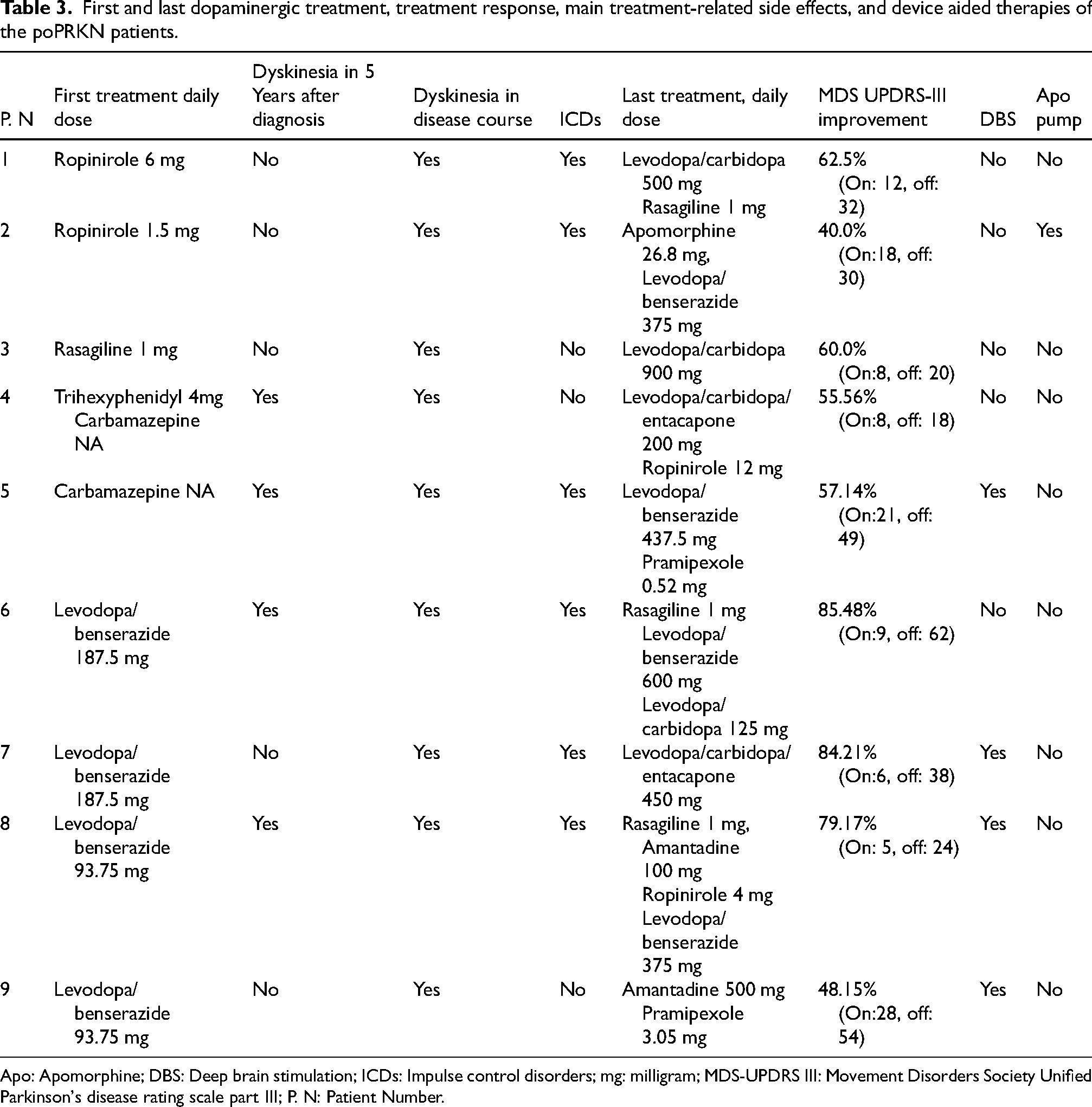
Apo: Apomorphine; DBS: Deep brain stimulation; ICDs: Impulse control disorders; mg: milligram; MDS-UPDRS III: Movement Disorders Society Unified Parkinson's disease rating scale part III; P. N: Patient Number.
The first treatment was L-dopa in four patients (44%), a dopamine agonist in two patients (22%), carbamazepine in two patients (22%), rasagiline in one patient (11%), and trihexyphenidyl in one patient (11%).
The initial response to L-dopa was excellent, as indicated by the improvement in the MDS-UPDRS part III scores shown in Table 3. The mean percentage of the MDS-UPDRS part III score improvement was 63.58% ± 16.06% (median: 60%, range: 40% to 84.21%). L-dopa was eventually started in all patients. The mean time until the start of L-dopa treatment since the initial diagnosis was 3.22 ± 3.18 years (median; 2, range; 1–10). Dyskinesia occurred in four patients (44%) within the first year of L-dopa treatment, and in three patients (33%) during the third year of the L-dopa treatment. All patients eventually developed dyskinesia as the disease progressed, even if the time of onset was very variable (up to 15 years since the L-dopa initiation; see Table 3). Strategies to manage dyskinesia included lowering the L-dopa daily dose (nine patients), partially or completely replacing L-dopa with dopamine agonists (four patients), using apomorphine pump (one patient), adding amantadine (three patients) or clonazepam (two patients), or undergoing surgery (four patients). The latter consisted in bilateral subthalamic nucleus (STN) deep brain stimulation (DBS) in four patients whereas bilateral globus pallidus internus (GPi) DBS was done in one patient.
Six patients (66%) developed impulse control disorders (ICDs) as a treatment-related side effect. Gambling was reported in four patients (patients 1, 5, 7, 8), hypersexuality in four patients (patients 1, 2, 4, 5), compulsive shopping in three patients (patients 1, 5, 8), and risky behavior in one patient (patient 6). As indicated, the same patient often presented with different types of ICD. ICDs developed under ropinirole in three patients (patients 1, 2, 8), pergolide in one patient (patient 7, who developed ICD a second time under pramipexole), ropinirole plus L-dopa in one patient (patient 6), and pramipexole plus apomorphine in one patient (patient 5). The primary strategy for managing ICDs was to lower or stop the dopamine agonist, and to start or increase the dose of L-dopa. Additionally, STN DBS was beneficial for reducing dopaminergic therapy in three patients.
Specifically concerning invasive treatment, five patients (55%) required device-aided therapy. Four received bilateral STN DBS, one of whom additionally underwent bilateral GPi DBS. DBS was performed at 8, 16, 21, and 24 years after disease onset (patient 5, 7, 8, and 9, respectively). GPi DBS was applied 41 years after disease onset in patient 9. Further details about DBS therapy can be found in Tables 3 and 4 and in the Supplemental Material.
Decrease in the MDS-UPDRS part III scores, and LEDD after DBS treatment.

DBS: Deep brain stimulation; GPi: Globus pallidus interna; LEDD: Levodopa Equivalent Daily Dose; P.N: Patient Number; MDS-UPDRS III: Movement Disorders Society Unified Parkinson's disease rating scale part III; STN: subthalamic nucleus.
The mean percentage of decreases in the MDS-UPDRS part III scores after STN DBS was 41.42% ± 22.64% (median: 43.25%, range: 12.50% - 66.67%). DBS was beneficial for motor symptom and dyskinesia management, leading to a mean percentage of reduction of the levodopa equivalent daily dose of 46.17% ± 20.00% (median: 45.24%, range: 25.70% - 68.52%) after surgery. (See details in Table 4). GPi DBS was beneficial for dyskinesia management (further details are available in the Supplemental Material). One patient received apomorphine pump therapy 22 years after disease onset (see the Supplemental Material for further details).
Discussion
Although PRKN-related PD is the most common cause of PD with pediatric-onset, prompt diagnosis seems to remain a relevant issue.6,11,16,22,23 Moreover, in the pediatric population, detailed clinical features of PRKN-related PD are lacking. From a non-systematic review of the available literature, most studies have focused solely on young-onset or juvenile PRKN-related PD, and lack of comprehensive clinical data (Table 5). A few studies have included poPRKN in their cohorts14,16,21 but without specific focus or detailed discussion on this specific population. Therefore, our study adds relevant data to the current available literature.
Main clinical characteristics of PRKN patients available in the literature and poPRKN patients of the present study.
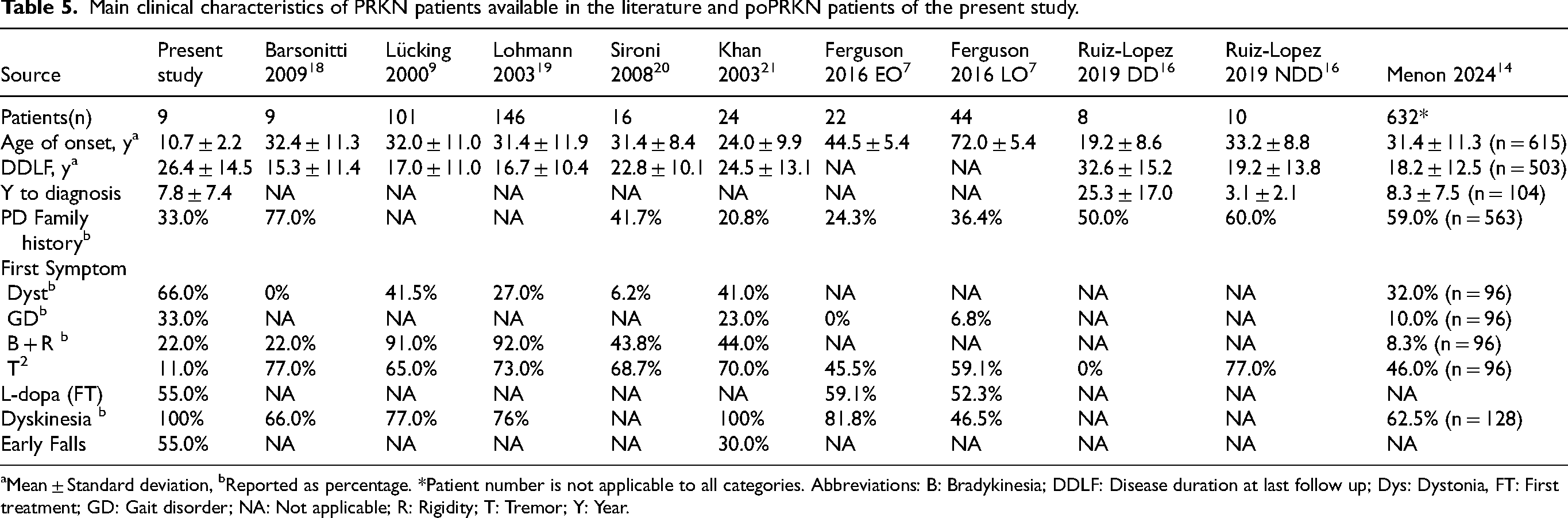
Mean ± Standard deviation, bReported as percentage. *Patient number is not applicable to all categories. Abbreviations: B: Bradykinesia; DDLF: Disease duration at last follow up; Dys: Dystonia, FT: First treatment; GD: Gait disorder; NA: Not applicable; R: Rigidity; T: Tremor; Y: Year.
The impact of the type of PRKN variant (missense, frameshift), the copy number variants, and the variant's location within the gene (C-terminus/exons 7–12, middle/exons 3–6, or N-terminus/exons 1–2) on the age of onset is largely unknown, although there may be a role. 14 Patients with frameshift variants had an earlier age of onset. 14 Moreover, the age of onset decreased as the number of involved exons increased, with each additional exon contributing to an earlier onset. Additionally, variants located in exons encoding the N-terminus of the protein were associated with an earlier age of onset compared to variants located in the middle or C-terminus. 14 In our cohort, copy number variants were closer to the N-terminus of the protein in eight patients (either exon 2, 3, or 4).
Dystonia has been previously reported as the first symptom in 6.20% to 41.50% of all PRKN-related PD cases (Table 5).9,14,18–21 In our poPRKN population, the frequency was higher (66%), possibly related to the very young age of PD onset. Our results suggest to always consider poPRKN in the initial differential diagnosis of children with focal dystonia.
The available literature is limited regarding non-motor symptoms in PRKN-related PD patients.14,24 While sleep disturbances (72.50%), 14 psychological disturbances (64.5%) 14 and dysautonomia were frequently reported (60%),14,21 cognitive impairment was uncommon.14,19,21 Moreover, PRKN-related PD was not found to be particularly associated with any specific psychiatric finding.25,26 Similarly, in our patients, we observed high prevalence of sleep problems, anxiety, and depression. Intriguingly, we have observed that non-motor symptoms always appeared after the motor symptoms’ onset, and later within the course of the disease, in contrast with older age PD onset. This is a new observation that needs to be further explored.
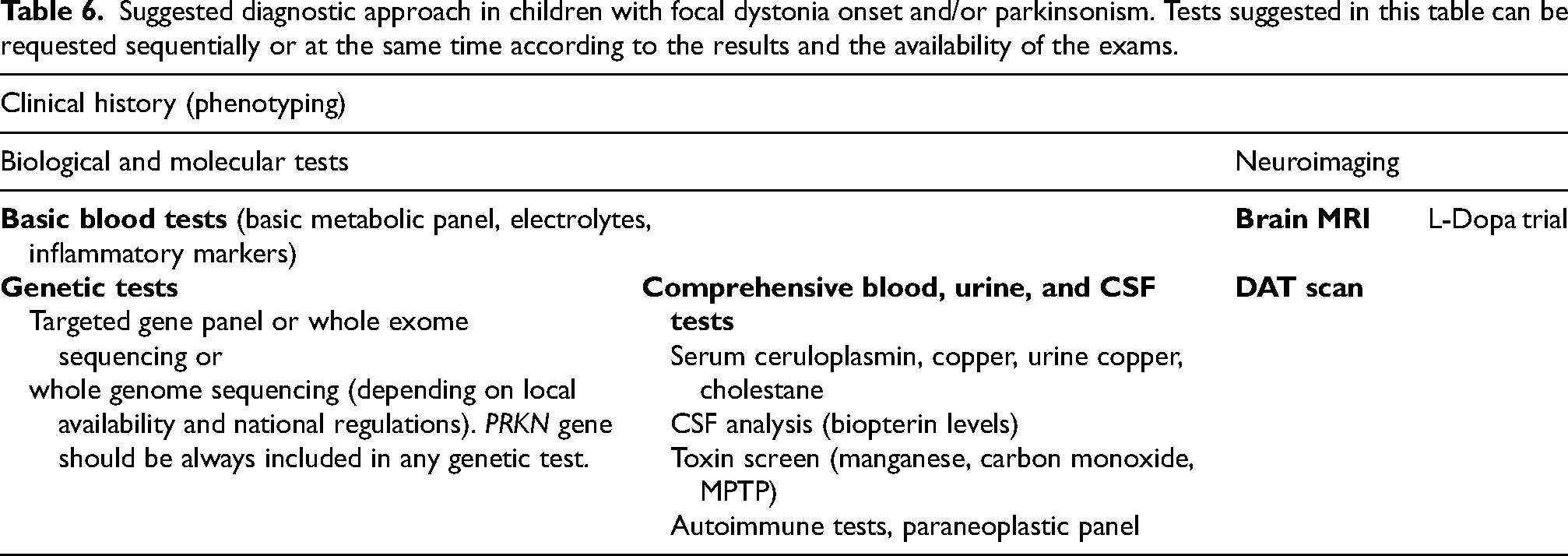
Early-onset PRKN-related PD patients are often misdiagnosed.27,28 Our study included six patients whose first symptom was dystonia, and three of them were initially diagnosed as dystonia. Diagnostic delay in non-poPRKN patients was previously evaluated only in one study. 16 This study compared the clinical characteristics of PRKN-related PD with delayed (25.30 ± 17.00 years) and non-delayed (3.10 ± 2.10 years) diagnosis. 16 The delayed diagnosis group had a younger age at disease onset (19.20 ± 8.60) while the non-delayed diagnosis group had an older age at disease onset (33.2 ± 8.8). 16 Our study, involving patients with a pediatric age of onset, further supports that a younger age at disease onset is associated with a delayed diagnosis. In our population, diagnosis of three patients was impressively delayed until when they saw an adult neurologist who made the correct diagnosis more than ten years after the disease onset (Table 1). All these findings suggest that disease awareness between pediatricians is crucial to overcome diagnostic delay. When poPRKN is suspected, while waiting for genetic testing results, a DAT-scan can be very useful to facilitate the correct diagnosis. Indeed, all our patients had positive DAT-scans like idiopathic PD. 29 In Table 6, we propose a rational approach for diagnosis of poPRKN.
Suggested diagnostic approach in children with focal dystonia onset and/or parkinsonism. Tests suggested in this table can be requested sequentially or at the same time according to the results and the availability of the exams.
PRKN-related PD respond excellently to L-dopa. 25 However, troublesome dyskinesia, even with low doses of L-dopa, is almost always reported in PRKN-related PD.21,25 Similarly, in our patients, the response to L-dopa was excellent, but in most patients, dyskinesia developed quickly within the first three years after started L-dopa, even with low doses (on average 120 mg/day). Our results are consistent with several previous studies reporting that the prevalence of dyskinesia tends to increase with a lower age of onset (Table 5).7,9,18,21,25 Conversely, a recent study reported a later onset of dyskinesia in PRKN-related PD, at around 19 years of disease duration, compared to EOPD (9.1 years). 14 The authors suggest that since PRKN-related PD often manifests during childhood or adolescence, the dopaminergic system can develop compensatory mechanisms that delay dyskinesia onset. 14 However, this study lacks information on the type of initial treatments, the timing of the first L-dopa initiation, the L-dopa daily dose, and when dyskinesias appeared post-treatment. In our study, the timing of the first L-dopa initiation and the dosage were specified, and dyskinesia rapidly appeared after the initiation of L-dopa, even with low daily dosage. Therefore, the late-onset dyskinesia reported in PRKN-related PD in the mentioned study 14 suffers from several biases that question the hypothesis of a developmental compensatory mechanisms of the dopaminergic system. Overall, considering the potential long-term complication of L-dopa, one treatment strategy can be to prioritize non-L-dopa treatment options as first line therapy. Nevertheless, L-dopa should be administered at the lowest doses able to satisfactory improve motor signs. On the other hand, the use of dopamine agonists in our patients was associated with frequent onset of ICDs. The use of dopamine agonist has been associated with a 2–3.5-fold increased risk of ICD in PD patients. 30 ICDs have also been linked to earlier disease onset, and longer disease duration.31,32 Although younger patients are more likely to be treated with dopamine agonist, the effect of age remains significant even after controlling for dopamine agonist exposure.30,33 Additionally, PRKN-related PD patients tend to have a higher frequency of ICDs compared to the general PD population. 34 The prevalence of ICDs in the general PD population ranges between 3.5%-43%, 32 and ICDs prevalence in PRKN-related PD was reported 21.4% and 68.18% in two previous studies.14,34 Therefore, poPRKN is also expected to be associated with a higher risk of ICDs, complicating the medical management of these patients. Taking all this into consideration, early DBS surgery might the best management of motor and non-motor complications linked to medical treatment. Four out of nine patients of our study underwent STN DBS surgery (Table 4), which led to better dyskinesia control, significant drug reduction, and marked improvement in the MDS-UPDRS part III scores.
Finally, while there is no clearly defined genotype-phenotype correlation, 25 some studies reported differences in olfactory disfunction between carriers of homozygous or heterozygous mutations in different exons.35,36 While our study population is relatively small to establish definitive conclusions, our findings indicate that the presence of heterozygous or homozygous mutations did not significantly influence the initial symptoms, motor or non-motor features, or treatment-related issues within the poPRKN population.
Our study has several limitations. The main limitation is the small number of patients. Additionally, the lack of standardized follow-ups due to the retrospective nature, and the diverse follow-up time (ranging between 6–56 years) were other limitations. Despite these limits, our study is the first study to comprehensively and specifically evaluate poPRKN patients within a long-follow-up.
Conclusions
The phenotype of pediatric onset PD caused by PRKN mutation has not been thoroughly described in the previously available literature. Atypical symptoms, such as foot dystonia or gait impairments, as initial presentations can markedly delay the diagnosis. Although the response to dopaminergic agents is very good, dyskinesia and ICDs are the most common, early-onset and disabling treatment-related side effects. Non-motor PD features appear rather later within the course of the disease. These finding suggest that genetic testing and DAT-scan should be performed as soon as genetic PD is suspected, and especially in a child with foot dystonia as initial clinical presentation. Moreover, dopaminergic treatment effects should be careful evaluated due to the frequent and early medication-related complications. DBS surgery is very effective in this population, and it should be early discussed and proposed when medical treatment side effects are not manageable.
Supplemental Material
sj-docx-1-pkn-10.1177_1877718X241296153 - Supplemental material for Pediatric-onset PRKN disease: New insights into an understudied population
Supplemental material, sj-docx-1-pkn-10.1177_1877718X241296153 for Pediatric-onset PRKN disease: New insights into an understudied population by Ozge Gonul Oner, Céline Biboulet Bruneau, Valérie Fraix, Véronique Bourg, Luc Defebvre, Eugénie Mutez, Emmanuel Roze, Cécile Laroche, Matthieu Béreau, Marie-Ange Nguyen-Morel and Elena Moro in Journal of Parkinson's Disease
Footnotes
Acknowledgments
We thank the patients and families who participated in this study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
EM has received honoraria from Medtronic for consulting. She has also received research grant support from Merz, Grenoble Alpes University, and France Parkinson.
The remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.