
Abstract
Systemic sclerosis (SSc) is a rare autoimmune condition with complex pathogenesis characterized by a heterogeneous presentation and different disease courses. Fibrosis of multiple organs including the lungs favored by inflammation and vasculopathy is the hallmark of SSc. SSc-associated interstitial lung disease (SSc-ILD) is common and can be associated with poor outcomes, this complication being the leading cause of death in recent series. Because of its huge heterogeneity, SSc-ILD management can be very challenging. Immunosuppressive therapy has long been used to prevent SSc-ILD progression with modest effects in clinical trials. However, thanks to a better understating of SSc pathogenesis, innovative therapies including antifibrotics are increasingly being developed. The achievement of the Safety and Efficacy of Nintedanib in Systemic SClerosIS (SENSCIS) trial has led to the approval by drug agencies of the first antifibrotic drug for SSc-ILD. In parallel, other antifibrotics are being investigated as possible beneficial therapies in SSc-ILD. An important unmet need remains to clarify the positioning of the various strategies, such as the added value of combination of immunosuppressants and antifibrotic therapies in patients at high risk of progression. Indeed, irreversible lung injury or self-perpetuated progression highlights the concept of a window of opportunity in SSc-ILD patients. Herewith, we provide an overview of the most significant clinical trials with antifibrotic drugs developed in recent years for the management of SSc-ILD and a viewpoint about their positioning in treatment algorithms.
Introduction
Systemic sclerosis (SSc) is a chronic, debilitating multisystem connective tissue disorder characterized by small-vessel vasculopathy and immune system activation/dysregulation culminating in progressive fibrosis of the skin and internal organs. 1 Although the pathogenesis of SSc is extremely complex and only partly known, the interplay between both genetics and specific environmental agents are thought to play pivotal roles in SSc development. 2 Fibrogenesis subsequent to failed tissue repair processes resulting in irreversible scarring and organ failure is the end-result of different mechanisms involved in the pathogenesis of SSc. 3 The recruitment of inflammatory cells with predominant type-2 helper (Th2) T-cells, but also B cells and dendritic cells, decreased functional regulatory T-cells (Tregs) and the increased production of proinflammatory and profibrotic cytokines (interleukin-1 (IL-1), IL-6, IL-13, IL-4, and transforming growth factor-β (TGF-β)) contribute to the phenotypic differentiation of fibroblasts into myofibroblasts and extracellular matrix (ECM) overproduction.4,5 One of the consequences of this inflammatory cascade leading to fibrosis is the development of interstitial lung disease (ILD). Indeed, ILD is common in SSc and is associated with poor outcomes. It has been identified as the leading cause of death in patients with SSc in recent series.6,7 Estimates of the prevalence of ILD in SSc vary depending on how ILD is defined. Nevertheless, the reported prevalence of ILD on high-resolution chest computed tomography (CT) scan (HRCT) varies from 22% and 50% according to reports from different large SSc cohorts.8–13 SSc-associated interstitial lung disease (SSc-ILD) shows similarities to idiopathic pulmonary fibrosis (IPF) in its natural disease course but tends to progress more slowly and to have a better prognosis. 14 By contrast with IPF, ILD pattern in SSc has more frequently a histologic/imaging pattern of nonspecific interstitial pneumonia (NSIP) rather than usual interstitial pneumonia (UIP). 15 Although lung involvement patterns in SSc are very heterogeneous, around 30% of patients with SSc-ILD experience ILD progression.16,17
The disease’s impact is compounded by a failure to identify therapeutic targets that reverse the natural history of the disease. However, as the understating of the pathogenesis of this heterogeneous disease improves, innovative specific therapies such as antifibrotics are increasingly being developed. The achievement of the Safety and Efficacy of Nintedanib in Systemic SClerosIS (SENSCIS) trial has led to the approval by drug agencies of the first antifibrotic drug for SSc-ILD. 18 Notwithstanding, despite the progress made in developing therapeutic options in the last few years, efforts are still needed to identify pharmacological strategies that can control disease progression and antifibrotics seem to be promising candidates. We searched PubMed and ClinicalTrials.gov for studies with antifibrotic drugs in patients with SSc (149 and 11 references identified, respectively). Nine relevant clinical trials mainly focusing on SSc-ILD have been identified and included in this narrative review. A summary of the clinical trials included are described in Table 1.
Summary of the most recent clinical trials with antifibrotics in SSc-ILD.
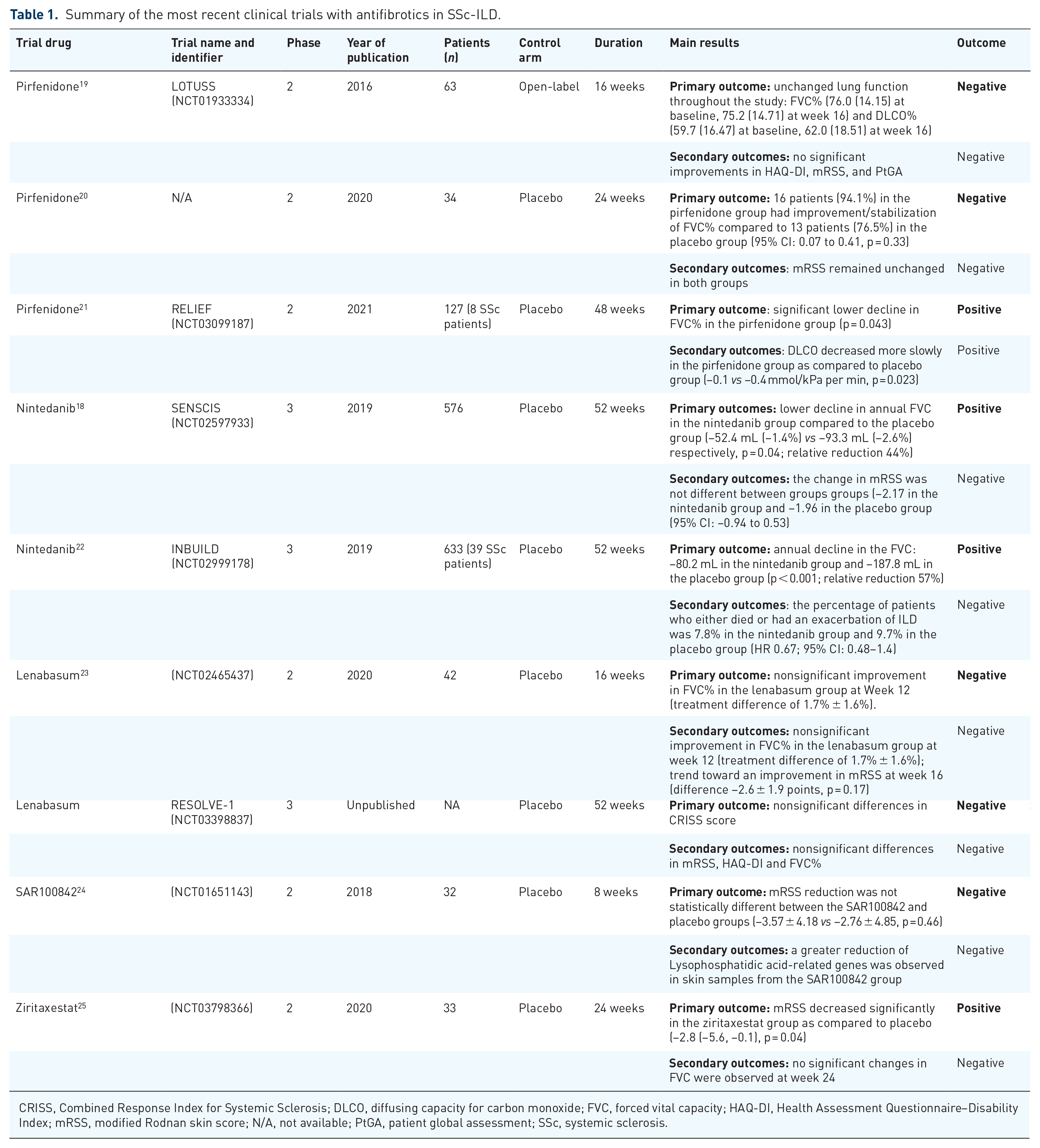
CRISS, Combined Response Index for Systemic Sclerosis; DLCO, diffusing capacity for carbon monoxide; FVC, forced vital capacity; HAQ-DI, Health Assessment Questionnaire–Disability Index; mRSS, modified Rodnan skin score; N/A, not available; PtGA, patient global assessment; SSc, systemic sclerosis.
Pirfenidone (in 2011) and nintedanib (in 2014) have been approved by regulatory agencies for the treatment of patients with IPF and are available for clinical use worldwide. The approval of these two antifibrotic drugs led to an increasing interest in their development for other non-IPF progressive ILD conditions including SSc-ILD. Therefore, we start by reviewing the major studies that lead to the approval of these drugs in IPF.
Antifibrotics in IPF
IPF is a chronic, progressive lung disease with a dismal prognosis and a median survival of 3–5 years from diagnosis.26,27 Advances in the mechanistic understanding of IPF pathogenesis and the development of antifibrotic drugs are changing the natural course of this condition. 28
While the precise mechanism of action of pirfenidone is not completely understood, it seems to exhibit both anti-inflammatory and antifibrotic effects. Its molecular mechanism is thought to involve the suppression of TGF-β reducing collagen synthesis and deposition. 29 On the contrary, nintedanib is a small-molecule tyrosine kinase inhibitor (TKI) which binds to adenosine triphosphate (ATP) blocking the downstream signaling of kinases including platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), and vascular endothelial growth factor receptor (VEGFR). 30
Pirfenidone has been evaluated for the treatment of IPF in two pivotal phase-3, multinational, randomized, controlled trials (CAPACITY studies 004 and 006), showing that it significantly reduced the decline in forced vital capacity % (FVC) at week 72 in study 004, but not in study 006.31,32 To further investigate the effects of pirfenidone (2403 mg per day) on disease progression in patients with IPF the Assessment of Pirfenidone to Confirm Efficacy and Safety in IPF (ASCEND) study was performed. 33 This was a phase-3, multicentre, randomized, placebo-controlled trial, that included 555 patients with IPF. The primary endpoint was the change from baseline to week 52 in the percentage of the predicted FVC. Treatment with pirfenidone resulted in a significant reduction of 48% in the proportion of patients who had a decline of ⩾10% of FVC or who had died (p < 0.001). The mean decline in FVC was 235 mL in the pirfenidone group and 428 mL in the placebo group (relative reduction 45%, p < 0.001). The proportion of patients with no decline in FVC% was increased by 132% in the pirfenidone group (63 patients (22.7%) vs 27 patients (9.7%) Pirfenidone reduced the relative risk of death or disease progression by 43% (HR, 0.57; 95% CI: 0.43−0.77; p < 0.001). The authors observed lower all-cause mortality in the pirfenidone group (11 (4.0%) vs 20 (7.2%)) although this difference did not reach significance (HR, 0.55; 95% CI: 0.26–1.15; p = 0.10). Regarding tolerability, gastrointestinal (nausea) and skin toxicity were more frequent in patients treated with pirfenidone; however, these AEs were generally mild to moderate. Based on the findings of the ASCEND trial, worldwide drug agencies approved pirfenidone for the treatment of patients with IPF.
Regarding nintedanib, a phase-2 randomized, placebo-controlled trial (TOMORROW trial) showed promising results suggesting that 12 months of treatment with nintedanib (150 mg twice daily) was associated with a reduced decline in FVC, fewer acute exacerbations, and the preservation of health-related quality of life. 34 To confirm these results two phase-3 trials (INPULSIS-1 and INPULSIS-2) were conducted. 35 In these trials, 1066 patients with IPF and a FVC of ⩾50% and a diffusing capacity of the lungs for carbon monoxide (DLCO) between 30% and 80% were included. The primary endpoint was the absolute annual rate of decline in FVC. In both trials, the annual rate of decline in FVC was significantly lower in the nintedanib group as compared to the placebo group (−114.7 mL vs −239.9 mL, p < 0.001, in INPULSIS-1 and −113.6 mL vs −207.3 mL, p < 0.001, in INPULSIS-2). In INPULSIS-2, there was a significant increase in time to first acute exacerbation in the nintedanib group (HR: 0.38; 95% CI: 0.19−0.77; p = 0.005) that was not observed in INPULSIS-1 (HR: 1.15; 95% CI: 0.54–2.42; p = 0.67). Furthermore, there was a trend toward a reduced mortality in the group of patients treated with nintedanib (5.5% in the nintedanib group and 7.8% in the placebo group (HR: 0.70; 95% CI: 0.43–1.12; p = 0.14). In both trials, the proportion of patients with serious AE was similar in the nintedanib and placebo groups. However, mild or moderate diarrhea was very frequent in patients treated with nintedanib (93.7% in INPULSIS-1% and 95.2% in INPULSIS-2) leading to premature discontinuation of the study drug in 14 patients in the nintedanib group (4.5%) and none of the patients in the placebo group in INPULSIS-1 and in 14 patients receiving nintedanib (4.3%) and 1 receiving placebo (0.5%) in INPULSIS-2. The results of INPULSIS-1 and INPULSIS-2 trials led to the approval of nintedanib for the treatment of patients with IPF.
Data from large registries assessing the course of disease of IPF patients also support the efficacy of antifibrotic therapy under real-life conditions. The results from the European IPF registry (eurIPFreg) on 525 IPF subjects recruited between 2009 until 2016 showed that steroids, immunosuppressants, and N-Acetylcysteine prescriptions declined since 2009 being progressively replaced by antifibrotics (pirfenidone and nintedanib). 36 Strikingly, when assessing survival in this registry, the median survival rates of patients taking antifibrotics were significantly higher than those treated with any other drugs (123.1 vs 68.3 months, p = 0.001). More recently, data from the German INSIGHTS-IPF registry showed that despite an overall decline in FVC and DLCO that was not significantly different between patients receiving or not antifibrotic therapy, the 1-year and 2-year survival rates were significantly higher in patients taking antifibrotic drugs (87% vs 46% and 62% vs 21%, respectively). 28 Indeed, the risk of death was 37% lower in patients taking antifibrotic therapies (HR: 0.63, 95% CI: 0.45−0.87).
Antifibrotics in SSc: pre-clinical data
Animal models exhibiting all the aspects of SSc are not currently available, they are nonetheless useful in providing clues into the understanding of the pathogenesis of this disease. 37
Treatment with pirfenidone attenuated fibrosis and had anti-inflammatory effects in the mouse model of bleomycin-induced pulmonary fibrosis. The effects observed with pirfenidone were mediated by the suppression of TGF-β gene expression 38 and downregulation of procollagen I and III expression 39 as well as suppression in proinflammatory cytokines IL-1, IL-6, IL-12, and monocyte chemoattract protein (MCP)-1) production 40 consequently decreasing pulmonary vascular permeability and decreasing influx of inflammatory cells (neutrophils, macrophages, and lymphocytes) in the lung. 41
In vivo investigations revealed antifibrotic and anti-inflammatory effects of nintedanib in animal models of SSc. In three different experimental models of SSc, Huang et al. 30 demonstrated that nintedanib inhibited fibroblast activation and myofibroblast accumulation and dose-dependently prevented bleomycin-induced skin fibrosis, ameliorated fibrosis in both chronic graft-versus host disease model and tight-skin 1 mice. In another model of early SSc, the authors assessed the histological and molecular effects of nintedanib on PAH. Nintedanib prevented and attenuated pulmonary vascular wall thickness (p < 0.001), and the number of occluded pulmonary vessels (0.001 ⩽ p < 0.01), in FOS-related antigen-2 (Fra2) transgenic mice. These effects were mediated through pulmonary vascular smooth muscle cells inhibition (0.01 ⩽ p < 0.05), normalization of the serum levels of VEGF (0.001 ⩽ p < 0.01) and impaired type-2 macrophages polarization of monocytes (0.01 ⩽ p < 0.05). 42
Antifibrotics in SSc: clinical data
Pirfenidone
The efficacy and safety of pirfenidone has been evaluated in SSc-ILD patients in an open-label, multicenter, randomized, phase-2 trial (An Open-Label, Phase-2 STUdy of the safety and Tolerability of Pirfenidone when Administered to Patients with Systemic Sclerosis–Related Interstitial Lung Disease (LOTUSS study)) (NCT01933334). 19 Exploratory disease outcomes included pulmonary function tests (PFTs) (FVC and DLCO), Health Assessment Questionnaire–Disability Index (HAQ-DI), patient’s global assessment of disease activity (PtGA) and the modified Rodnan Skin Score (mRSS) at week 16. Overall, 63 SSc-ILD patients (mean age 50.6 years, 82.5% female, 49.2% diffuse type, 63.5% receiving background mycophenolate mofetil (MMF)), were assigned to receive pirfenidone 267 mg oral capsules at a starting dose of 801 mg/day (1 capsule, 3 × daily) titrated to a maintenance dose of 2403 mg/daily. FVC% (76.0 (14.15) at baseline, 75.2 (14.71) at week 16) and DLCO% (59.7 (16.47) at baseline, 62.0 (18.51) at week 16) remained unchanged throughout the study. Subgroup analysis of patients on background MMF at randomization showed that mean change from baseline in FVC% was 0.6% and −0.3% in the MMF and no-MMF subgroups, respectively; mean change from baseline DLCO% was 3.2% and −0.2% in the MMF and no-MMF subgroups at week 16, respectively. Moreover, the authors did not observe any significant improvements in other exploratory disease outcomes in particular HAQ-DI, mRSS (11.4 (9.61) at baseline, 11.2 (9.90) at week 16) and PtGA. Regarding safety assessments, the most common AE were nausea, headache, and fatigue. The majority of AE were mild to moderate; however, 19% of patients reported severe AE including fatigue (5%), diarrhea (3%), and nausea (3%). Since the different outcome measures remained unchanged throughout the trial probably related to the short follow-up design of this study, no conclusion of the efficacy of pirfenidone could be drawn.
A recent double-blind, randomized, placebo-controlled trial assessed the safety and efficacy of pirfenidone in SSc-ILD patients. 20 A total of 34 subjects (median age 41 years, 91% female, 35% diffuse type, 62% on background immunosuppressants) were randomized to receive either pirfenidone (2400 mg/day) (n = 17) or placebo (n = 17). The primary outcome was the proportion of patients with an absolute increase of FVC% ⩾ 10% or stabilization of FVC defined as an absolute change in FVC < 10% at 6 months. Sixteen patients (94.1%) in the pirfenidone group had improvement/stabilization of FVC% compared to 13 patients (76.5%) in the placebo group (95% CI: 0.07−0.41, p = 0.33). The median change in FVC% was −0.55% (−9% to 7%) and 1.0% (−42% to 11.5%) (p = 0.51) in the pirfenidone and placebo groups, respectively. In parallel, regarding extrapulmonary outcome measures, skin scores (mRSS) remained unchanged in both groups (0.0 vs −1.0, p = 0.26, in pirfenidone and placebo groups, respectively). In this trial, the most common AE were nausea, diarrhea, malaise, and skin rashes, but no significant differences were noted between the study groups.
A multicenter US-based phase-2 randomized controlled trial (Scleroderma Lung Study (SLS) III) assessing the efficacy and additive effects of a combination of MMF and pirfenidone has been conducted (NCT03221257) but was prematurely stopped before the complete enrolment of the expected number of patients because of the sponsor decision.
Very recently, pirfenidone has been evaluated in patients with progressive fibrotic ILD other than IPF (RELIEF trial) (NCT03099187). 21 In this phase-2, randomized, double-blind, placebo-controlled trial, the efficacy of pirfenidone was assessed in patients with ILD due to connective tissue diseases (CTDs), fibrotic non-specific interstitial pneumonia, chronic hypersensitivity pneumonitis, or asbestos-induced lung fibrosis, with FVC 40–90%, DLCO 10–90%, and an annual decline of FVC of at least 5% predicted despite conventional therapy. The primary endpoint was absolute change in FVC% from baseline to week 48. A total of 127 patients were randomized. The most common diagnosis were chronic hypersensitivity pneumonitis (57 patients (45%)), followed by CTDs (37 patients (29%)). Within the CTD-ILD group, the most common condition was rheumatoid arthritis (17 patients (46%)). Eight patients (22%) had SSc. Overall, 73% and 89% of patients were taking glucocorticoids or immunosuppressant therapy in the pirfenidone and placebo groups, respectively. Concerning the primary endpoint, a significant lower decline in FVC% in the pirfenidone group in the rank ANCOVA analysis (p = 0.043) was observed. At week 48, absolute change in FVC was -37 and -114mL in pirfenidone and placebo groups respectively, (p = 0.21). On the contrary, DLCO decreased more slowly in the pirfenidone group as compared to placebo group (−0.1 vs −0.4 mmol/kPa per min, p = 0.023). There were five deaths in the placebo group (8%) and one death in the pirfenidone group (2%). However, no significant difference was found between pirfenidone and placebo with regard to progression-free survival. Similarly, no significant difference was noted between-group regarding 6-minute walking distance and quality of life. The most frequent serious AE were infections (5 (8%) in the pirfenidone group and 10 (16%) in the placebo group). Serious AEs were observed but rare: nausea (two patients in the pirfenidone group and two patients in the placebo group), dyspnea (one patient in the pirfenidone group and one patient in the placebo group) and diarrhea (one patient in the pirfenidone group). These data suggest that treatment with pirfenidone might modestly attenuate disease progression in patients with progressive fibrotic ILD other than IPF.
Nintedanib
The efficacy and safety of nintedanib in SSc-ILD were assessed in a phase-3 trial (Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) trial (NCT02597933) 18 which is the largest trial ever performed in SSc. A total of 576 SSc-ILD patients (mean age 54 years, 52% diffuse type) with a disease duration <7 years (the median time since the onset of the first non-Raynaud’s symptoms was 3 years) were randomized to receive either 150 mg of nintedanib orally twice daily or placebo. Patients receiving low-dose prednisone and/or stable background therapy with MMF or methotrexate were allowed to participate. ILD was defined based on HRCT showing fibrosis affecting at least 10% of the lungs, and FVC ⩾ 40% and DLCO between 30% and 89%. Key exclusion criteria included clinically significant pulmonary hypertension and >3 digital ulcers or history of severe digital necrosis requiring hospitalization. The primary endpoint was the annual rate of absolute decline in FVC over a 52-week period. The annual decline in FVC was lower in the nintedanib group compared to the placebo group (−52.4 mL (−1.4%) vs −93.3 mL (-2.6%) respectively, relative reduction 44%; p = 0.04). Regarding secondary outcome measures, the change in mRSS was not different between groups (−2.17 in the nintedanib group and −1.96 in the placebo group (95% CI; −0.94 to 0.53)). Changes in FVC decline were not reflected in an improvement in health-related quality of life as HAQ-DI was not significantly different between nintedanib and placebo groups. Moreover, mortality was similar between the two groups with 10 deaths (3.5%) and 9 deaths (3.1%) in nintedanib and placebo groups, respectively.
The annual decline in FVC among patients who were receiving MMF at baseline (48% of the included patients) was −40.2 mL in the nintedanib group and −66.5 mL in the placebo group (relative reduction 40%) as compared to −63.9 and −119.3 mL (relative reduction 46%), respectively in patients who were not receiving MMF. Despite the limitations in comparing groups of patients who had not undergone randomization according to baseline MMF therapy, these encouraging data suggest a potential benefit of MMF in association with nintedanib on lung function.
Regarding safety, the most common AE was diarrhea which was observed in 75.7% of the patients in the nintedanib group (vs 31.6% in the placebo group). AE leading to treatment discontinuation were higher in the nintedanib group than in the placebo group (16.0% vs 8.7%). On the contrary, the percentage of patients with any serious AE was similar between both groups.
Based on the results observed on patients on stable treatment with MMF, a post hoc subgroup analysis of the SENSCIS trial was performed aiming to examine the efficacy and safety of nintedanib by MMF use at baseline. 43 The proportion of patients with an absolute decrease in FVC of at least 3.3% predicted at week 52 was analyzed. This post hoc analysis showed that the proportion of patients with a decrease in FVC ⩾ 3.3% was lower with nintedanib than with placebo in both patients on background MMF (29% vs 40% of patients; OR 0.61 (0.37–1.01)) and not on MMF at baseline (40% vs 47% of patients; OR 0.73 (0.46–1.16)). The authors found no heterogeneity in the effect of nintedanib on the annual rate of decline in FVC between the subgroups by MMF use (p value for interaction = 0.45). Furthermore, the AE profile of nintedanib was similar between the subgroups. These findings suggest that the combination of MMF and nintedanib might be a safe and efficacious treatment option for patients with SSc-ILD. Nevertheless, further data are needed on the benefits of initial combination therapy compared to a sequential approach in patients with SSc-ILD.
Based on the results of the SENSCIS trial, nintedanib was granted by several drug agencies for the treatment of SSc-ILD.
The results of the SENSCIS trial were completed by the INBUILD trial (NCT02999178) assessing nintedanib in miscellaneous progressive fibrosing ILD. 22 In this phase-3 trial, patients with fibrosing ILD were assigned to receive nintedanib (150 mg twice daily) or placebo. All patients had ILD affecting more than 10% of lung volume on HRCT, FVC ⩾ 45% and DLCO comprised between 30% and 80%. Progressive ILD was defined as follows: a relative decline in the FVC% of at least 10%, a relative decline in the FVC% of 5% to less than 10% and worsening of respiratory symptoms or an increased extent of fibrosis on HRCT. Background immunosuppressants at inclusion were not allowed. The primary endpoint was the annual absolute decline in the FVC. An enrichment strategy was used with randomization being stratified according to the fibrotic pattern (two thirds of the patients had a UIP-like fibrotic pattern). A total of 663 patients (mean age 65.8 ± 9.8 years, 54% men, FVC% at baseline was 69.0 ± 15.6%) were included. Thirty-nine patients (5.9%) had SSc-ILD. Other diagnosis included hypersensitivity pneumonitis (26%), rheumatoid arthritis-associated ILD (13%) and mixed CTD-associated ILD (3%). In the overall population, the decline in the FVC was −80.2 mL per year in the nintedanib group and −187.8 mL per year in the placebo group (p < 0.001; relative reduction 57%). The difference in the annual decline in the FVC was even more marked in patients with a UIP-like pattern: −82.9mL per year in the nintedanib group and −211.1 mL in the placebo group (p < 0.001; relative reduction 61%). At week 52, the percentage of patients who either died or had an exacerbation of ILD was 7.8% in the nintedanib group and 9.7% in the placebo group (HR 0.67; 95% CI, 0.48–1.4). As reported in the SENSCIS trial, diarrhea was the most frequent AE (66.9% in the nintedanib group vs 23.9% in the placebo group). However, the percentages of patients with any AE and serious AE were similar in the nintedanib group and the placebo group.
Other antifibrotic drugs
Lenabasum
The endocannabinoid pathway abrogates the activation of endothelial cells acting as modulator of endothelial/inflammatory cell interaction. 44 Furthermore, cannabinoids have immunomodulatory effects on the activation and proliferation of T- and B-cells, macrophages, as well as proinflammatory and profibrotic cytokines.45,46 Cannabinoid receptors in particular CB2 are also present in skin fibroblasts and their activation prevented skin fibrosis in different preclinical models of SSc.47,48 Lenabasum is an oral agonist of CB2 that activates the resolution phase of innate immune responses and exerts antifibrotic effects. 49
Lenabasum reduced TGF-β and collagen production by fibroblasts improving skin and lung fibrosis in experimental models of SSc.50–52 Based on these data, a phase-2, multicenter, randomized, double-blind, placebo-controlled trial was conducted (NCT02465437). 23 A total of 42 patients with <6 years’ duration dcSSc were randomized in an overall 2:1 ratio of lenabasum or placebo. Patients treated with lenabasum showed greater improvement in the CRISS score at week 16 (primary outcome) as compared to placebo (0.33 (interquartile range (IQR) 0.01−0.82) vs 0.00 (IQR: 0.000−0.16), p = 0.07). A trend toward an improvement in mRSS at week 16 was observed in the lenabasum group compared to the placebo group (difference −2.6 ± 1.9 points, p = 0.17). Exploratory lung assessments were performed showing a non-significant numerical improvement in FVC% in the lenabasum group at week 12 (treatment difference of 1.7% ± 1.6%). AE occurred in 63% of the lenabasum group and 60% in the placebo group and no serious AE related to lenabasum were observed. Subjects who completed the 16-week period phase-2 trial were eligible to receive lenabasum (n = 36) in an open-label extension (21 months) (congress data). 53 The CRISS median score continued to improve (0.96 (IQR 0.43) at week 93 compared to 0.33 at week 16) and mRSS declined by −10.3 ± 7.2 points. FVC% decreased 3.2% from study start. The excellent safety profile of lenabasum observed in the 16-week period was confirmed during the open-label extension phase. Despite these encouraging data, the preliminary data of the phase-3 RESOLVE-I trial (NCT03398837) showed no significant differences in the primary (CRISS score) and secondary endpoints (mRSS, HAQ-DI, FVC%) at 1 year compared to placebo.
Lysophosphatidic acid axis antagonists
Lysophosphatidic acid (LPA) is a lipid mediator which is converted from lysophosphatidylcholine through the lysophospholipase activity of autotaxin that plays key pathogenic role in fibroblast recruitment and activation, vascular leak, and endothelial dysfunction. In a preclinical mouse model of pulmonary fibrosis the absence of LPA receptors led to reduced fibroblast recruitment and vascular leak protecting the experimental mice from fibrosis and mortality. 54
Supporting data showed a plausible role in IPF. Indeed, a phase-2, randomized, double-blind, placebo-controlled trial of an LPA receptor 1 antagonist (BMS-986020) was performed in patients with IPF (FVC 45%–90%; DLCO 30%–80%). 55 The primary endpoint was the absolute change in FVC from baseline to week 26. Overall, 143 patients were randomized and 108 completed the 26-week dosing phase. The authors observed that patients treated with BMS-986020 showed a slower decline in FVC as compared to placebo (−42 mL (95% CI: −116 to −22) vs -13 mL (−201 to −68), respectively; p = 0.049). Three cases of cholecystitis related to BMS-986020 lead to premature study discontinuation. Two phase-3 trials assessing the efficacy of GLPG1690 (ziritaxestat), an autotaxin-LPA axis antagonist in patients with IPF are currently ongoing (NCT03711162 and NCT03733444).
Recent studies have also suggested that the LPA axis plays an important role in the pathogenesis of SSc.56–59 An 8-week, phase-2, double-blind, placebo-controlled trial assessing the effects of SAR100842, a potent-selective LPA receptor 1 antagonist, in patients with early (less than 36 months) dcSSc (NCT01651143). 24 SAR100842 at 300 mg or matching placebo was administered orally twice a day. Immunosuppressive therapies prior to enrolment were permitted. This trial was followed by an open-label uncontrolled 16-week extension phase of the study with the same dosage of SAR100842. The primary endpoint was safety and tolerability during the 8-week period and secondary endpoints were change in mRSS and change in blood and skin biomarkers from baseline to week 8. Thirty-two patients participated in the blinded 8-week period and 30 participated in the open-label extension study. The safety profile of SAR100842 was acceptable during the blinded and open-label periods of the study. Most treatment-related AE were mild to moderate (80% in the SAR100842 group vs 71% in the placebo group). At week 8, the mRSS reduction was not statistically different between the SAR100842 and placebo groups (−3.57 ± 4.18 vs −2.76 ± 4.85, p = 0.46). A greater reduction of LPA-related genes was observed in skin samples from the SAR100842 group. However, after 24 weeks of treatment, patients treated with SAR100842 showed a clinically meaningful decrease in mRSS (mean ± SD change −7.36 ± 4.24; median change −7.50). Unfortunately, no efficacy assessments on lung function were performed.
More recently, ziritaxestat, an autotaxin-LPA axis antagonist was investigated in patients with dcSSc in a phase-2, randomized, double-blind, placebo-controlled trial (NCT03798366). 25 The primary endpoint was change in mRSS at 24 weeks. Secondary endpoints included safety and tolerability and changes in FVC. Thirty-three patients were randomized (mean age 49.3 years, 69.7% female). Even though not clinically meaningful, mRSS decreased significantly in the ziritaxestat group as compared to placebo (–2.8 (–5.6, −0.1), p = 0.04). No significant changes in FVC were observed at week 24. Ziritaxestat was well tolerated, and most AEs were mild or moderate.
These results support a possible role for the LPA axis in the treatment of SSc; however, further clinical research is warranted.
Figure 1 illustrates the pathogenesis of SSc-ILD highlighting the recent antifibrotic agents described above.
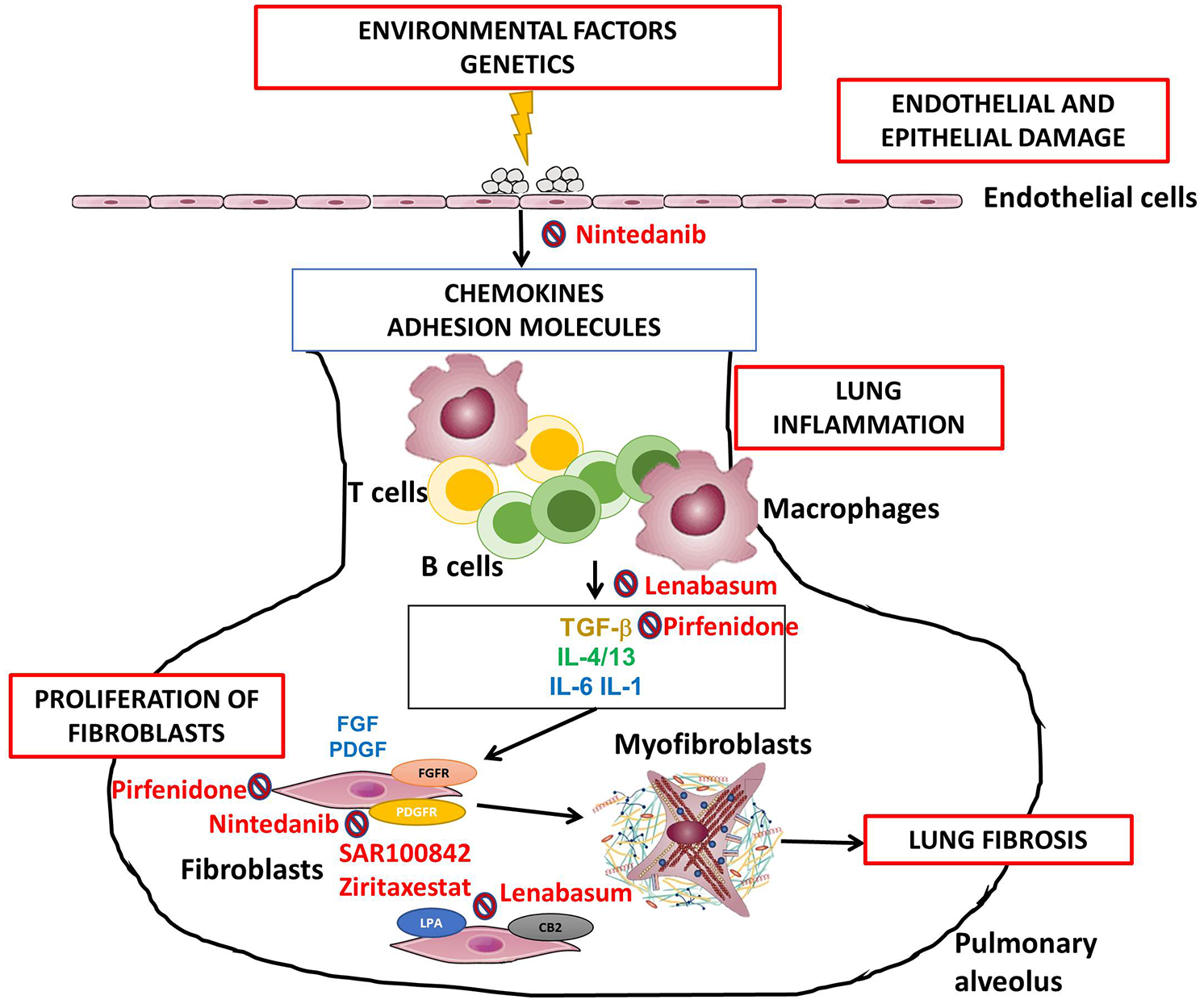
A model of systemic sclerosis-associated interstitial lung disease pathogenesis highlighting recent antifibrotic agents. Legend: vascular and epithelial injury results in an increased production of chemokines and adhesion molecules leading to the recruitment and accumulation of leucocytes and activation and proliferation of B-cells, T-cells, and macrophages. The secretion of profibrotic and proinflammatory cytokines causes activation and differentiation of resident fibroblasts into myofibroblasts. Myofibroblasts organize and contract the extracellular matrix leading excessive lung fibrosis. Cellular and molecular therapeutic targets are indicated. TGF-β, transforming growth factor-β; IL, interleukin; LPA, lysophosphatidic acid receptor; CB2, cannabinoid receptor type 2; FGF, fibroblast growth factor; PDGF, platelet-derived growth factor; FGFR, fibroblast growth factor receptor; PDGFR, platelet-derived growth factor receptor.
Practical considerations
Notwithstanding the recent approval of nintedanib for the management of SSc-ILD, a formal consensus on the use of antifibrotic drugs regarding treatment initiation, escalation, and patient selection is still lacking. Nevertheless, different panel of experts have issued a number of viewpoints to help physicians to better understand the positioning of antifibrotics in SSc-ILD.60,61 The following preliminary considerations have been put forward. Thanks to systematic screening, early diagnosis is common in SSc; however, heterogeneity is very common in FVC trajectories with about one third of patients being progressive. SENSCIS inclusion criteria must be carefully analyzed in any SSc-ILD patient candidate to drug therapy. The first step is to split infra-clinical (asymptomatic patients, marginal extent and normal or close to normal PFTs) and clinical ILD patients. So far, only clinical ILD may be treated with antifibrotic therapies based on SENSCIS criteria. The challenging question for clinical ILD is whether antifibrotic drugs should be given first or in escalation when progression is observed despite first line immunosuppressants. Guidance may come from the inflammatory status of the disease and also extrapulmonary findings. Intuitively, predominant lung disease in a patient free of systemic inflammation might be treated first by antifibrotics whereas active inflammatory disease (high CRP), the presence of extrapulmonary activity (skin progression, arthritis, tendon friction rubs, and myositis), and auto-antibody status in particular anti-topoisomerase I antibodies might guide one toward immunosuppressants first. Disease duration seems also of great importance and probably patients with early disease might benefit from immunosuppressants first. One unanswered question in SSc-ILD is whether ILD HRCT pattern should be taken into consideration; the interpretation of fibrotic NSIP is challenging, and further insight may come in the future from more detailed HRCT analyses including artificial intelligence and/or potential biomarkers. Regardless of the first line option, tight follow-up will be crucial: indeed, progressive patients might benefit from step-up combination therapy including antifibrotics and immunosuppressants. A consensus on the definition of progression will be helpful for practical management of SSc-ILD patients and the following has been suggested: progressive symptoms with increased lesions in HRCT, a relative decline of ⩾10% in FVC, relative decline in FVC of 5% or more with decline in DLCO of 15% or more, or a relative decline of ⩾5% FVC with progressive symptoms or increased lesions in HRCT over 24 months despite treatment. 62 Table 2 shows factors that should be taken into consideration by physicians when choosing between immunosuppressants or antifibrotics in SSc-ILD.
Summary of potential factors to be taken into consideration when choosing between immunosuppressants or antifibrotics in SSc-ILD.

HRCT, high-resolution computed tomography; ILD, interstitial lung disease; MSK, musculoskeletal; SSc, systemic sclerosis.
Conclusion
Despite the variable course of SSc-ILD and overall better survival outcomes than IPF, disease progression is common, and timely effective treatment approaches are critical for preserving or slowing the decline in lung function.17,62,63 Moreover, recent preclinical data showed that lung injury can cause non-resolving pulmonary fibrosis through mechanical impairment of lung microenvironment thus suggesting a therapeutic window of opportunity in early SSc-ILD. 64
Until recent years, standard-of-care treatment options in SSc-ILD were limited to conventional immunosuppressants. 61 However, the recent advent of the SENSCIS trial opened the therapeutic avenue for antifibrotics in the management of SSc-ILD. One of the greatest unanswered questions is that of the combination of immunosuppressants and antifibrotics in SSc-ILD. Up to now, the majority of clinical trials have investigated immunosuppressive or antifibrotic therapies in isolation rather than in combination. The subgroup analysis of the SENSCIS trial provided some data on the additive effect of a classic immunosuppressant (MMF) and nintedanib. Furthermore, tocilizumab has recently been the first biologic immunosuppressant approved by the Food and Drug Administration for the treatment of SSc-ILD. 65 The promising potential of an upfront combination of targeted immunosuppressants and antifibrotic therapies especially in patients at high risk of severe disease progression refractory to conventional immunosuppressants, still lacks evidence but appears as a very appealing future strategy to improve the outcomes of a dreadful disease.
Footnotes
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GB and JA declare no conflict of interests relevant to the content of this article. YA reports personal fees from Actelion, Bayer, BMS, Boehringer, Curzion, Inventiva, Roche and Sanofi and research grants from Inventiva, Sanofi and Alpine Immunosciences.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.