
Abstract
Background:
The aim of this study will be to investigate the therapeutic effect and safety of non-steroidal anti-inflammatory drugs (NSAIDs) along with symptomatic slow-acting drugs for the treatment of osteoarthritis (SYSADOA), JOINS tablets, for degenerative knee osteoarthritis (OA) treatment and to determine the analgesic and anti-inflammatory effects of the combination therapy. In addition, we will investigate whether JOINS treatment alone after NSAID and JOINS combination treatment is effective in relieving and maintaining knee OA symptoms.
Methods:
This study will be a prospective, randomized, double-blind endpoint study design. All patients will be randomly assigned to either intervention (celecoxib+JOINS) or control (celecoxib+placebo) groups. In Part 1, the intervention group will be administered celecoxib once a day and JOINS three times a day for a total of 12 weeks. In the control group, celecoxib will be administered once a day and JOINS placebo three times a day for a total of 12 weeks. In Part 2, JOINS alone and JOINS placebo alone will be administered for an additional 24 weeks in both groups, respectively. The primary endpoint will be the amount of change during the 12 weeks as assessed using the Western Ontario and McMaster Universities Osteoarthritis Index total score compared with baseline. The secondary endpoint will be the amount of change at 1, 4, 12, 24, and 36 weeks from the baseline for pain visual analog scale, Brief Pain Inventory, Short Form Health Survey-36 and biomarkers.
Results:
The trial was registered with Clinical-Trials.gov (NCT04718649). The clinical trial was also registered on Clinical Research Information Service (CRIS) with the trial registration number KCT0005742.
Conclusions:
The combination treatment of the most commonly used SYSADOA drug, JOINS, and selective COX-2 inhibitor celecoxib as the representative NSAID for knee OA treatment, can be compared with celecoxib alone treatment to determine the safety or therapeutic effect.
Keywords
Introduction
Osteoarthritis (OA) is the most common joint disorder and knee OA is the most common location.1–4 Knee OA is clinically indicated by knee pain, stiffness, restriction of joint motion, and reduced function due to disintegration of the knee cartilage.5,6 The goal of treatment is to reduce pain, improve joint movement, and prevent further damage to the joint. 7 Various methods are used as conservative treatments of knee OA, but the most representative pharmacological treatments are non-steroidal anti-inflammatory drugs (NSAIDs) and analgesics.8–10 NSAIDs are effective drugs for knee OA symptom relief, but various side effects including gastrointestinal toxicity have been reported.11,12 Compared with traditional NSAIDs, selective cyclooxygenase-2 (COX-2) inhibitor celecoxib, which theoretically reduces gastrointestinal adverse events (AEs), has been developed but it is still controversial in terms of safety.13,14 In addition, although these NSAIDs are effective in reducing pain and inflammation, they cannot be designated as a fundamental treatment for knee OA. 15 Accordingly, interest in symptomatic slow-acting drugs for osteoarthritis (SYSADOA) is gradually increasing. 16
SYSADOA used in the treatment of knee OA include glucosamine, chondroitin, and diacerein.17–20 In many studies, SYSADOA had a similar pain reduction effect to NSAIDs, and thus could reduce the use of NSAIDs.17–20 In addition, there have been studies showing that a combination of SYSADOA and NSAIDs is more effective than a single treatment alone.20–22 In Korea, SYSADOA is used in 43% of knee OA patients, 23 and JOINS is a representative SYSADOA drug.16,23,24 Several studies have suggested that JOINS can prevent degradation of OA in vitro,25–27 but in vivo data are sparse. 24 In a study comparing JOINS with NSAIDs, a randomized controlled trial (RCT) showed cartilage protection, but there were limitations to analysis due to the small number of samples. 24 In addition, there have been no studies on the effects of combined treatment with SYSADOA and NSAIDs.
JOINS tablet (SK Chemicals, Seongnam, Korea) is a herbal anti-arthritic drug that has various physiological activities and is a new concept for OA treatment. 24 The JOINS tablet contains powder extracts of medicinal herbs including Clematis mandshurica, Trichosanthes kirilowii, and Prunella vulgaris mixed in an alcoholic aqueous solution at a 1:2:1 weight ratio, then fractionated with water-saturated butanol, and the residual solvents are completely removed. 24 JOINS had anti-inflammatory, analgesic, and joint cartilage protection effects in in vitro studies.25–27 JOINS tablets have been shown to improve articular cartilage metabolism and might delay the progression of degenerative OA in the knee and compensate for the disadvantages of the selective COX-2 inhibitor.24–27 SYSADOA, in combination with NSAID, is more effective in improving knee pain and function in knee OA compared with drug monotherapy.20–22 It is hypothesized that if celecoxib and JOINS tablet are used together, it might exert synergistic effects on the treatment of degenerative arthritis of the knee. 20 Therefore, the aim of this study is to investigate the therapeutic effect and safety of NSAID with JOINS tablet treatment for degenerative knee OA and to determine the analgesic and anti-inflammatory effects of the combination therapy. In addition, the effect of JOINS monotherapy in maintaining symptoms after NSAID and JOINS combination treatment will be investigated.
Methods
Patient population
All patients will meet the following criteria to be eligible:
Inclusion criteria
Men and women of at least 50 years of age.
Patients will be followed in an ambulatory clinic.
Patients presenting with primary OA of the knee according to American College of Rheumatology criteria. 28
Patients with OA of radiographic stages 2 or 3 according to the Kellgren–Lawrence grade.
Patients with knee pain on most days for at least 6 months with a visual analog scale (VAS) pain score (0–10) while walking on a flat surface ⩾4.
Patients with no clinically significant laboratory abnormalities based on judgment of the investigator.
Patients agreeing to sign informed consent prior to any study-related activities after having been clearly informed of its methods and constraints.
Patients not taking part in any other clinical study.
Patients agreeing to respect the protocol by attending all visits related to the study.
Subjects with one or more of the following conditions will be excluded:
Exclusion criteria
Patients with secondary knee OA (inflammatory arthritis including gout, reactive arthritis, rheumatoid arthritis, psoriasis, psoriatic arthritis, and traumatic OA).
Patients with known hypersensitivity to herbal products, hypersensitivity to celecoxib, who demonstrated allergic-type reactions to sulfonamides, experienced asthma, urticaria or allergic-type reactions after taking sulfonamides, aspirin [acetyl salicylic acid (ASA)], lactose, NSAIDs, acetaminophen, or paracetamol.
Patients with other bone and articular diseases (antecedents and/or current signs) such as chondrocalcinosis, Paget’s disease of the ipsilateral limb to the target knee, rheumatoid arthritis, aseptic osteonecrosis, gout, septic arthritis, ochronosis, acromegaly, haemochromatosis, Wilson’s disease, osteochondromatosis, seronegative spondyloarthropathy, mixed connective tissue disease, collagen vascular disease, psoriasis, and inflammatory bowel disease.
Patients with fibromyalgia.
Patients who will undergo surgery in any lower limb or arthroscopy, aspiration, or lavage in any lower limb joint within 180 days of the screening visit.
Patients who underwent previous knee replacement or surgery including arthroscopy on the study knee.
Patients who will undergo total knee replacement in the contralateral knee within 180 days prior to the screening visit.
Patients with co-morbid conditions or joint deformity (valgus and varus deformity >30° based on the Hip-Knee-Ankle angle) 29 that restrict knee function.
Patients with a history of heart attack or stroke, or who had serious diseases of the heart such as congestive heart failure (functional classes II–IV of the New York Heart Association ).
Patients with poorly controlled diabetes mellitus defined as hemoglobin A1c level >8%.
Patients with poorly controlled hypertension (sustained systolic blood pressure of >150 mmHg or diastolic blood pressure >95 mmHg).
Patients with any active acute or chronic infections requiring antimicrobial therapy, or serious viral (e.g., hepatitis, herpes zoster, HIV positivity) or fungal infections.
Patients with a history of recurrent upper gastrointestinal (UGI) ulceration or active inflammatory bowel disease (e.g., Crohn’s disease or ulcerative colitis), a significant coagulation defect, or any other condition, which in the investigator’s opinion might preclude the chronic use of celecoxib or diacerein. Patients may, at the investigator’s discretion, take a proton pump inhibitor (PPI) or antacids daily as required, with a 2-h period between intake of study medication and intake of PPI or antacid.
Patients with chronic liver or kidney disease, as defined by aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >2.0 times the upper limit of normal (ULN) or blood urea nitrogen (BUN) or serum creatinine >2.0 times ULN, at the screening visit.
Patients with any clinically significant conditions such as active cancer, severe respiratory, neurological or cerebral diseases.
Patients with a history of intolerance to acetaminophen or paracetamol, opioids or opioid combinations such that an adequate non-anti-inflammatory rescue analgesic regimen cannot be safely prescribed.
Patients who plan for any surgery during the study.
Patients with the impossibility of taking part in the total duration of the study and attending the visits.
Patients receiving intraarticular injections of hyaluronic acid or corticosteroids within 3 months prior to the screening visit.
Patients who will have injected or systemically administered corticosteroids within 3 months prior to the screening visit (systemic administration: daily oral corticosteroids or inhalation exceeding 1500 µg).
Genetic problems such as galactose intolerance (related to celecoxib), Lapp lactose deficiency, or glucose-galactose malabsorption.
Pregnant or lactating women.
This study will be performed at one single university hospital.
Randomization and blinding
This study will be a prospective, randomized, double-blind endpoint study design. One investigator in charge of patient allocation will be selected and will generate a randomization table. The list of assignments will be kept by the Clinical Trials Pharmacy (CTP) in a password-protected file. After randomization, all patients will be randomly assigned to either the intervention group (celecoxib + JOINS) or control group (celecoxib + placebo) with a distribution ratio of 2:1 (144:72). The investigator in charge of allocation will not intervene in any other process of this study and will participate only in the task of selecting an allocation group using a random number table. The evaluation will be conducted by another investigator who is not aware of patient allocation by the randomization table. Investigators will determine the treatment for each participant by requesting a CTP. However, the procedure for violating this regulation during treatment will only be performed in medical emergencies when treatment relies on knowledge of the drugs actually received. If the treatment code for the participant is broken, or if any other action such as a record date or reason for breaking blindness, discontinuation of clinical treatment will be reported to the sponsor.
Intervention
According to the inclusion and exclusion criteria, patients who participate in the study will be randomly assigned to two groups. The ratio between the intervention group and the control group will be 2:1. This study consists of two parts (Part 1 and 2). In Part 1, the intervention group will be administered 200 mg celecoxib (Celebrex, SK Chemicals, Seongnam, Korea) once a day and 200 mg of JOINS three times a day for a total of 12 weeks. In the control group, 200 mg of celecoxib will be taken once a day and JOINS placebo three times a day for a total of 12 weeks. The placebo tablet has the same size, color, shape and texture as JOINS, and contains sugar, corn starch, and microcrystalline cellulose. In Part 2, celecoxib will be stopped in patients who successfully completed Part 1. In the intervention group, JOINS alone monotherapy will be continued for an additional 24 weeks, and in the control group, JOINS placebo alone will continue for 24 weeks. 30 Patients will visit the outpatient clinic five times for a screening examination at 4 weeks, 12 weeks, 24 weeks, and 36 weeks. The whole study will be conducted out of the outpatient clinic.
Drugs will be distributed to patients three times over a total of 36 weeks during the study period. First, 12 weeks of medication will be distributed at the start, followed by 12 weeks of medication at the 12th and 24th weeks. If the patient medication is not be able to control the pain and additional medication is requested, Tylenol 650 mg will be allowed as a rescue drug to be taken up to three times a day during the follow-up period, and the use of the rescue medication will be recorded (Figures 1 and 2).

Flow chart for the treatment of knee osteoarthritis.
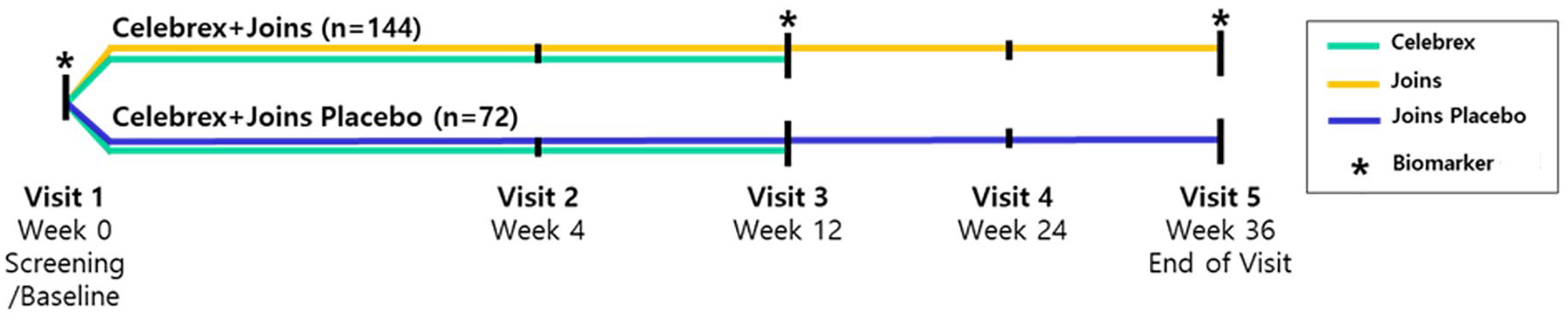
Schematic scheme.
Outcome measures
Baseline data
The baseline data, including gender, age, patient-reported height (cm) and weight (kg), family status, highest reached level of education, duration of OA pain symptoms, Kellgren–Lawrence grade, 31 number of painful joint/body regions, comorbidities, smoking and alcohol consumption, pain medication consumption, presence of psychological disease such as depression or anxiety, and type and duration of analgesic used before study will be collected. In general, knee X-rays will be taken at each outpatient visit and knee degenerative arthritis patients will be measured for joint space width (JSW) 32 and Kellgren–Lawrence grade 31 to objectively evaluate the efficacy and stability of the medication effect. The total patient evaluation period is 36 weeks. The schedule of measurements is presented in Table 1.
Schematic timeline.

Primary outcome
Primary endpoint will be the amount of change in the 12-week Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total score compared with the baseline. 33 The WOMAC is the most commonly used and validated assessment tool for evaluating the treatment effects of knee OA. The WOMAC consists of pain, stiffness, and function, with five items for pain, two items for stiffness, and 17 items for function, totaling 24 items. Each question is assigned a score of 0 (none) to 4 (extreme) on a 5-point Likert scale. Therefore, the total score ranges from 0 to 96 points, and the higher the score, the more inferior the clinical outcome. WOMAC will be measured at 4, 12, 24 and 36 weeks. 33
Secondary outcome
The secondary endpoint will be the amount of change at 1, 4, 12, 24, and 36 weeks from the baseline for pain VAS, Brief Pain Inventory (BPI), WOMAC function and Short Form Health Survey-36 (SF-36). The patient global impression of change scale, biomarkers, treatment compliance and blinding efficacy will also be evaluated. All AEs that occurred during the follow-up period will also be recorded.
Pain VAS
The patient pain score will be measured at resting, walking, and nighttime VAS using a 10-point Likert scale. A score of 0 is no pain and 10 was extreme pain. Resting, activity, and nighttime VAS will be tested at 4, 12, 24, and 36 weeks. 34
BPI
The BPI is a patient-reported scale used to measure pain severity and pain interference at 4, 12, 24, and 36 weeks after taking the drug. Pain severity will be assessed on four items, including the most severe pain, the least severe pain, the average pain over the last 24 hours, and the current pain. The pain scale uses a 10-point Likert scale that ranges from 0 (no pain) to 10 (extreme pain). Pain interference evaluates the impact of pain on seven items over the past 24 hours, including daily activities, mood, walking ability, ability to work normally, relationships with others, sleep, and enjoyment of life. Interference levels range from 0 (no interference) to 10 (complete interference). 35
Physical function assessment
WOMAC total score will be the primary outcome, and WOMAC function score will be used for physical function assessment; total WOMAC score ranges from 0 to 68 points. 33
SF-36
SF-36 is a short form of health status questionnaire made up of 36 questions consisting of eight different items, including physical function, social function, role-emotional, role-physical, physical pain, general health, mental health and vitality. This can be largely combined into two summary scores, the physical and mental health summary scores. For each summary score, the ordinal score is converted to a linear 0–100 scale where 0 represents the most unfavorable condition and 100 represents the best condition. 36
The patient global impression of change scale
The patient global impression of change (PGIC) scale is a standardized self-measurement tool that measures the change in overall subject condition after administration of an investigational drug. It is a 7-point scale and is divided into very much worse = −3, much worse = −2, slightly worse = −1, no change = 0, a little improved = +1, much improved = +2, very much improved = +3. The PGIC will be assessed at all visits after initiation of treatment. 37
Biomarkers
There are many biomarker research results that can confirm the extent of OA exacerbation.38,39 Among various biomarkers closely related to disease progression, C-terminal telopeptide of type II collagen (CTX-2), serum hyaluronan (HA), and N-telopeptides of type I collagen (NTX-1) are the most predictive models, and C-terminal telopeptide of type I collagen (CTX-1) and CTX-2 are the models that best show baseline status.38,39 In addition, serum cartilage oligomeric matrix protein (COMP) is also commonly used as a biomarker. Bone turn-over markers include CTX-1 and NTX-1, and cartilage turn-over markers include COMP, HA, and CTX-II. In general, these biomarkers are known to increase with disease progression.38,39 Therefore, it is possible to confirm the therapeutic effect of the drug more clearly through such biomarker tests. The biomarkers used in this study will be selected by referring to previous reports.38,39 All biomarkers will be assessed from serum samples, and the biomarker confirmation cycle will be measured before administration at 12 weeks and 36 weeks. 40
Adverse events
Any adverse reactions that may have occurred related to drug use during the study will be thoroughly documented.
Treatment compliance
Whenever the clinical trial drug is provided, the clinical investigator (or the person who has been delegated) is instructed to take medication so that the subject’s compliance can be maintained. Treatment compliance is evaluated based on the number of unused investigational drugs returned by the subject at the end of drug administration. To confirm compliance with the clinical trial drug, the investigator measures the quantity of the drug returned by the clinical study subject, calculates the actual dose, and checks the drug compliance as follows.
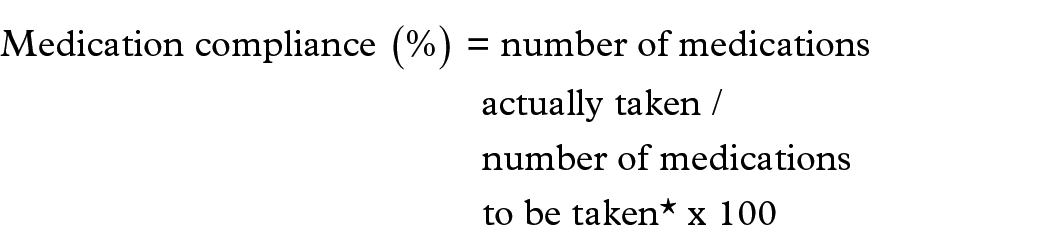
*Number of drugs to be taken: The total number of drugs to be taken from the time of prescription to the time of examination on the end of drug administration.
Treatment compliance should be at least 80% of the investigational medicinal product provided. To evaluate treatment compliance, the investigator (or delegated person) must record the quantity of the investigational drug provided and returned at each visit and the date on which the investigational drug was stopped. Subjects must receive medication instruction on the schedule for taking the investigational drug. 41
The blinding efficacy
A method to determine the effectiveness of blinding is to use a blinding questionnaire administered to the subject at the end of the trial. A questionnaire with three response categories will be used, including “new treatment,” “placebo (or control),” or “don’t know (DK)” for the treatment guess. The James Blinding Index (BBI) will be used to assess the blinding efficacy of the trial. 42
Sample size calculation
The basis for calculating the sample size calculation includes (1) prediction standard deviation: 19.0, (2) difference in the amount of change between the two prediction groups (test group–control group): −8.36, (3) level of significance, α = 0.05 (both sides), (4) Type 2 error (β) = 0.20, so the power is maintained at 80%.43–45 Due to the lack of clinical data for this test drug, the number of subjects will be calculated by referring to the literature on OA clinical trials of selective COX-2 inhibitors, which are widely prescribed for the treatment of OA symptoms. Based on the results of two previous similar studies, the difference in WOMAC total score change of −8.36 between the celecoxib and placebo groups at week 12 from baseline will be set as the difference in predicted change in this study.43,45 Since the standard deviation for the amount of change is not presented in the relevant document, the standard deviation of the predicted standard deviation is 19.0, referring to the standard deviation of 14.1–19.1 in similar literature that presented the standard deviation for the difference in the amount of change in the WOMAC total score at week 12 compared with the baseline. 44 After substituting different independent t-tests and calculating by setting the ratio of the intervention group to the control group to 2:1 using the G power program (Heinrich Hein University, Dusseldorf, Germany), 130 patients in the intervention group and 65 patients in the control group will be required. When the follow-up failure rate is considered at 10%, 144 patients in the intervention group and 72 people in the control group will be required; therefore, a total of 216 patients will be required for this study.
Statistical analysis
All data will be statistically analyzed using SPSS 24.0 software (IBM, Armonk, NY, USA) and followed the intention-to treat principle. Normally distributed measurement data will be expressed as the mean ± standard deviation. Non-normally distributed measurement data will be expressed as lower quartile (q1), median, and upper quartile (q3). Categorical data will be expressed as percentage. Repeated measures analysis of variance will be used for comparisons of WOMAC score for outcome measures between pre-treatment and 12, 24, and 36 weeks after treatment. An independent t-test of differences will be used for comparisons of WOMAC score and other outcome measures among the intervention and control groups at the same time point. Pearson’s chi-square test will be used for comparison of the incidence of AEs among the two groups at the same time point. An α = 0.05 (two-sided) will be used for inspection level.
Patient and public involvement
Neither the patients nor the public will be involved in the planning and design of this study.
Adverse events and data safety monitoring
All treatment-related AEs reported by patients during the study period will be recorded and evaluated at each visit. The safety and tolerability of treatment will be evaluated according to the incidence and type of AE. A treatment-related AEs will be defined as any event that occurs for the first time or worsens during treatment compared with the baseline when it first started. All AEs that the investigator determines to have a causal relationship with the study drug will be recorded using the case report form (CRF). Serious AEs will be reported to the Research Ethics Committee as soon as possible. The Research Ethics Committee will review all AEs and decide whether to continue or terminate the study. If a patient is harmed by participating in this clinical trial, the researchers treat the issue without delay and closely monitor the situation until resolved.
Ethical Considerations
This study has been approved by the Institutional Ethics Committee of Seoul Saint Mary’s Hospital (approval number: KC20MISV0634). The study will be conducted in accordance with the principles of the latest Helsinki Declaration Medical Research Act involving human subjects (WMO) and Good Clinical Practice (GCP) standards.
Conflict of interest
This work was supported by a research grant from SK Chemicals. The funder provides drugs, funding for research assistants/statisticians, and transportation expenses for participants. Participants in the study received 40 US dollars in the name of transportation expenses for each visit. None of the authors received reimbursements or salary from SK Chemicals. None of the authors hold any stocks or shares.
Dissemination
There is no agreement between the parties and investigators regarding the disclosure of research data. Research results will be presented at national or international scientific conferences and published in international peer-reviewed scientific journals. Patient data will be provided anonymously in all publications and presentations of the data.
Trial status
The trial is ongoing and is currently recruiting patients. Recruitment was initiated on February 10, 2021 and is expected to be completed by the end of February 2023. The trial was registered with Clinical-Trials.gov (NCT04718649) on January 22, 2021. The clinical trial was also registered on Clinical Research Information Service (CRIS) with the trial registration number KCT0005742 on the January 7, 2021.
Data monitoring
The Data Monitoring Committee (DMC) reviews patient data collected as patient recruitment progresses and checks safety independently of the Institutional Ethics Committee. 46 The DMC consists of a statistician, two research doctors, and an independent chair without any competing interests. The DMC will oversee and manage data integrity, security and efficiency, guide blind primary data analysis, and report research findings.
Data management
Personal data will be handled confidentially. 46 All participant data will be anonymized by assigning a unique participant identification code that includes an abbreviation of the study name followed by a sequence number. The unique participant identification list will be used to link the data to the participant. Data stored on the computer will be kept secure and password protected. All data will be entered electronically using an electronic CRF built on REDCap. Access to data will be provided to investigators and will be limited by user identifiers and passwords. Data will be analyzed using SPSS version 24.0 (SPSS Inc., Chicago, IL, USA) for statistical analysis.
Auditing
The Contract Research Organization will audit the trial conduct every 6 months independently of investigators and the sponsor, and will evaluate compliance with the protocol and the principles of GCP, and decide on any premature closure of the study. 46
Ancillary and post-trial care
Participants can be in touch with the researcher during the study period or up to 1 month after the end of the participation. 46 This study cannot guarantee the absence of unexpected and serious side effects during and after participation. Serious AEs will be addressed appropriately and promptly even after study completion.
The Catholic University of Korea has insurance to compensate for inadvertent damage related to the protocol. In addition, the Principal Investigator has purchased additional insurance to compensate for any damage to participants. Research insurance policies do not cover incidents that are believed to have occurred due to negligence, including incidents due to violations of important protocols.
Discussion
Celecoxib is an effective and commonly used NSAID for the treatment of knee OA. 14 Since it is currently the most widely used treatment worldwide, celecoxib was chosen from among the NSAIDs, and the default dose will be 200 mg per day. 14 This treatment period of combination treatment was based on a previous large celecoxib-related RCT that generally set 12 weeks as the treatment period.18,47–49 Continuous treatment with JOINS for 24 weeks was selected based on previous studies that set the treatment period of SYSADOA to 6 months, 17 and in the case of JOINS, no safety concerns were observed even with continuous administration for 1 year. 24
This study has several important features. First of all, the combination treatment of the most commonly used SYSADOA drug, JOINS, and selective COX-2 inhibitor celecoxib as the representative NSAID for knee OA treatment, can be compared with celecoxib alone treatment to determine the safety or therapeutic effect. Second, it is possible to assess the safety and therapeutic effect of JOINS alone treatment by additionally proceeding with JOINS alone after celecoxib and JOINS combination treatment, and the effect of continuing JOINS monotherapy on NSAID treatment effect can also be investigated. Third, it is expected that the drug side effects of long-term JOINS drug use can be accurately identified through long-term drug use for 36 weeks. Finally, by analyzing the biomarkers associated with knee OA progression, the effect of JOINS on disease progression can be precisely investigated.
Supplemental Material
sj-doc-1-tab-10.1177_1759720X211024025 – Supplemental material for Efficacy and safety of celecoxib combined with JOINS in the treatment of degenerative knee osteoarthritis: study protocol of a randomized controlled trial
Supplemental material, sj-doc-1-tab-10.1177_1759720X211024025 for Efficacy and safety of celecoxib combined with JOINS in the treatment of degenerative knee osteoarthritis: study protocol of a randomized controlled trial by Man Soo Kim, In Jun Koh, Yong Gyu Sung, Dong Chul Park, Sung Cheol Yang and Yong In in Therapeutic Advances in Musculoskeletal Disease
Footnotes
Authors’ Note
This study was performed in Department of Orthopedic Surgery, Seoul St. Mary’s Hospital, The Catholic University of Korea.
Conflict of interest statement
The author(s) declared the following potential conflicts of interestwith respect to the research, authorship, and/or publication of this article: This work was supported by a research grant from SK Chemicals, Seongnam, Korea. The funder provides drugs, funding for research assistants/statisticians, and transportation expenses for participants. Participants in the study received 40 US dollars in the name of transportation expenses for each visit. None of the authors received reimbursements or salary from SK Chemicals. None of the authors hold any stocks or shares.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication: This study was supported in part by funding from SK Chemicals, Seongnam, Korea.
Ethical Committee Approval
Institutional Ethics Committee approval of Seoul St. Mary’s Hospital, the Catholic University of Korea Study (approval number: KC20MISV0634). The trial was registered with ![]() (NCT04718649). The clinical trial was also registered on Clinical Research Information Service (CRIS) with the trial registration number KCT0005742.
(NCT04718649). The clinical trial was also registered on Clinical Research Information Service (CRIS) with the trial registration number KCT0005742.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.