
Abstract
Raynaud’s phenomenon (RP) is relevant to the rheumatologist because it may signify an underlying connective tissue disease and also because it can be very challenging to treat, especially when it has progressed to digital ulceration or critical ischaemia. This review article discusses diagnosis (does this patient have an underlying connective tissue disease?), including the role for nailfold capillaroscopy, and treatment. Management of ‘uncomplicated’ RP is first described and then treatment of RP complicated by progression to digital ulceration or critical ischaemia, highlighting recent advances (including phosphodiesterase type 5 inhibition, and endothelin 1 receptor antagonism) and the evidence base underpinning these. Possible future therapies are briefly discussed.
Introduction
Raynaud’s phenomenon (RP) is an exaggeration of the normal physiological response to cold exposure or to emotional stress. Typically the extremities turn white (ischaemia) then blue (deoxygenation) then red (reperfusion). The reason that management of what is a vascular phenomenon is included in a journal of musculoskeletal disease is twofold:
Although the vast majority of RP is primary (idiopathic, PRP), RP can be secondary to a number of different conditions, including connective tissue disease. 1 Importantly, RP is the most common presenting feature of systemic sclerosis (SSc) and can precede its diagnosis by many years. Therefore RP can present a window of opportunity to rheumatologists for early diagnosis of an underlying connective tissue disease.
RP in patients with connective tissue disease, especially in those with SSc, can be very severe with a major impact on quality of life, sometimes progressing to digital ulceration or critical ischaemia (sometimes to gangrene requiring amputation).2,3 In the order of 40–50% of patients with SSc will have at least one digital ulcer during the course of their illness.3,4 This is in contrast to the situation in patients with very common ‘benign’ PRP, which by definition does not progress to irreversible tissue injury. Management of connective tissue disease-associated RP can be very challenging.
This review article will consider the following:
Establishing the diagnosis in the patient presenting with RP, with specific reference as to whether or not there is an underlying connective tissue disease.
Treatment of ‘uncomplicated’ RP (RP which has not progressed to digital ulceration or critical ischaemia).
Treatment of RP which has progressed to digital ulceration or critical ischaemia.
Possible future therapies.
It should be emphasized that, in general, the evidence base for management of RP is weak. This reflects in part the difficulties in running randomized controlled trials (RCTs) 5 : usually these are time constricted to the winter months (to minimize the effects of the seasonality of ambient temperature) and a further complication is the lack of reliable outcome measures for RP (the only validated outcome measure is the Raynaud’s Condition Score, a patient-reported outcome). 6 However, things are changing, evidenced by several multicentre RCTs over the last 10 years. 7
Establishing the diagnosis
This is the first principle of management. RP is not in itself a diagnosis: it is a symptom complex requiring a diagnosis, which can usually be made with a combination of careful history and examination, backed up by some key investigations, looking specifically for any of the underlying conditions summarized in Table 1. If RP is primary,8,9 then the episodes or attacks should be entirely reversible, there should be no history of ulceration or gangrene, the peripheral pulses should be easily felt and symmetrical, there should be no evidence of digital pitting, ulceration or gangrene, the antinuclear antibody (ANA) should be negative or only weakly positive (titre < 1/100), the erythrocyte sedimentation rate (ESR) normal (although the newer Maverakis et al. criteria 9 do not require this) and the nailfold capillaries (examined via capillaroscopy) normal.
Main differential diagnosis of (and associations with) Raynaud’s phenomenon.
The rheumatologist is specifically interested in diagnosing an underlying connective tissue disease. Puffy fingers (on history and examination) are highly suspicious of early SSc.10,11 A number of other features on examination of the hands point towards SSc or a SSc spectrum disorder: sclerodactyly (sometimes in combination with more proximal skin thickening), digital pitting (Figure 1), dilated nailfolds (sometimes these are so dilated as to be visible to the naked eye, especially in patients with dermatomyositis) and telangiectases. Therefore careful examination of the hands is all important.

Digital pitting in a patient with systemic sclerosis.
The minimal set of investigations for a patient with RP (dictated by the criteria for PRP)8,9 comprises a blood count and ESR, ANA and nailfold capillaroscopy. Most rheumatologists would additionally request a biochemical profile with thyroid function, immunoglobulins with protein electrophoresis, and a chest or thoracic outlet radiograph (looking for a cervical rib). 12 Additional investigations depend on the index of suspicion as to what (if any) underlying disease might be present. If connective tissue disease is suspected, then more extensive immunology testing will be required, for example for SSc-specific autoantibodies (anticentromere, anti-topoisomerase, anti-RNA polymerase III). A SSc-specific autoantibody and abnormal nailfold capillaries are independent predictors for the development of SSc in a patient with RP.13,14 Specialist centres may have access to thermography and this too can help differentiate between PRP and SSc-related RP.
Nailfold capillaroscopy
At the nailfold, capillaries lie parallel (rather than perpendicular to) the skin surface and can be visualized noninvasively using the technique of nailfold capillaroscopy. Abnormal nailfold capillaries are an early manifestation of SSc10,11 providing the rationale for capillaroscopy as a key investigation in patients presenting with RP. Characteristic abnormalities include dilated capillaries, areas of avascularity, distortion of the normal nailfold architecture and haemorrhage15–17 (Figure 2). Abnormal nailfold capillaries are one of the 2013 American College of Rheumatology/European League Against Rheumatism (EULAR) classification criteria for SSc18,19 and it therefore behoves all rheumatologists making the diagnosis of SSc to be familiar with the technique of nailfold capillaroscopy and to have access to this; otherwise the diagnosis could be missed. Different capillaroscopic techniques are available. While the ‘gold standard’ is high magnification videocapillaroscopy (200–600×), there is increasing interest in lower magnification, ‘hand-held’ techniques, the dermatoscope,20–23 or (more recently) the USB microscope. These low-magnification techniques (and including the original widefield microscopy described by Maricq and Le Roy) 15 have the advantage of instantaneous imaging of the whole nailfold, facilitating easy and quick detection of abnormalities. While an opthalmoscope can also be used and allows detection of dilated capillaries, 20 in the author’s experience it is more difficult to visualize the capillaries clearly than with the dermatoscope or USB microscope.

(a) Normal and (b) abnormal nailfold capillaries, with dilated capillaries, distortion of the normal nailfold architecture and areas of avascularity.
Thermography
Infrared thermography allows indirect measurement of blood flow, by imaging surface temperature, and can help to differentiate between PRP and SSc-related RP.24–28 Although its use is currently confined to specialist centres, it is possible that the introduction of low-cost ‘mobile-phone’ systems may lead to increased application of thermography in the evaluation of RP. Standardizing protocols (for example, of cold challenge testing) across centres is required to encourage wider adoption of the technique.
Treatment of uncomplicated RP
As already stated, the evidence base for the treatment of RP (primary and secondary) is limited. The general approach to the management of RP is summarized in Figure 3, which is modified from the UK Scleroderma Study Group consensus best practice pathways. 12 The fact that the pathway already requires modification [with phosphodiesterase type 5 (PDE5) inhibitors ‘moving up’ the pathway], highlights how there have been significant recent advances in the management of RP over the last 5 years.

Modification of the UK Scleroderma Study Group best practice recommendations on the management of Raynaud’s phenomenon. 12 Phosphodiesterase inhibition has been ‘moved up’ the original pathway to be positioned along with other oral vasodilator therapies. Note that clinicians outside the UK might modify their approach depending on their access to therapies. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker: CCB, calcium channel blocker; PDE5, phosphodiesterase type 5; SSRI, selective serotonin reuptake inhibitor. Modified from Herrick. 7
Removal of any underlying cause
When applicable, this is always the first step. For example, removing vibration exposure in patients with hand–arm vibration syndrome or discontinuing any drug therapy (if possible) which might be aggravating RP.
General/lifestyle measures
Patients should try to avoid cold temperatures and should dress warmly (including warm gloves and socks). They should be strongly encouraged to stop smoking. Patient education is a key aspect of management and leaflets published by patient support organizations give very valuable information. For many patients (especially those with PRP), these measures will be sufficient to control symptoms: in a substantial proportion of patients, RP improves over time. 29
Drug treatment
Those patients not responding to general measures should be offered drug treatment (Table 2). This will apply to patients with severe PRP, to many of those with connective tissue disease related RP, and to most of those with SSc-related RP. 30
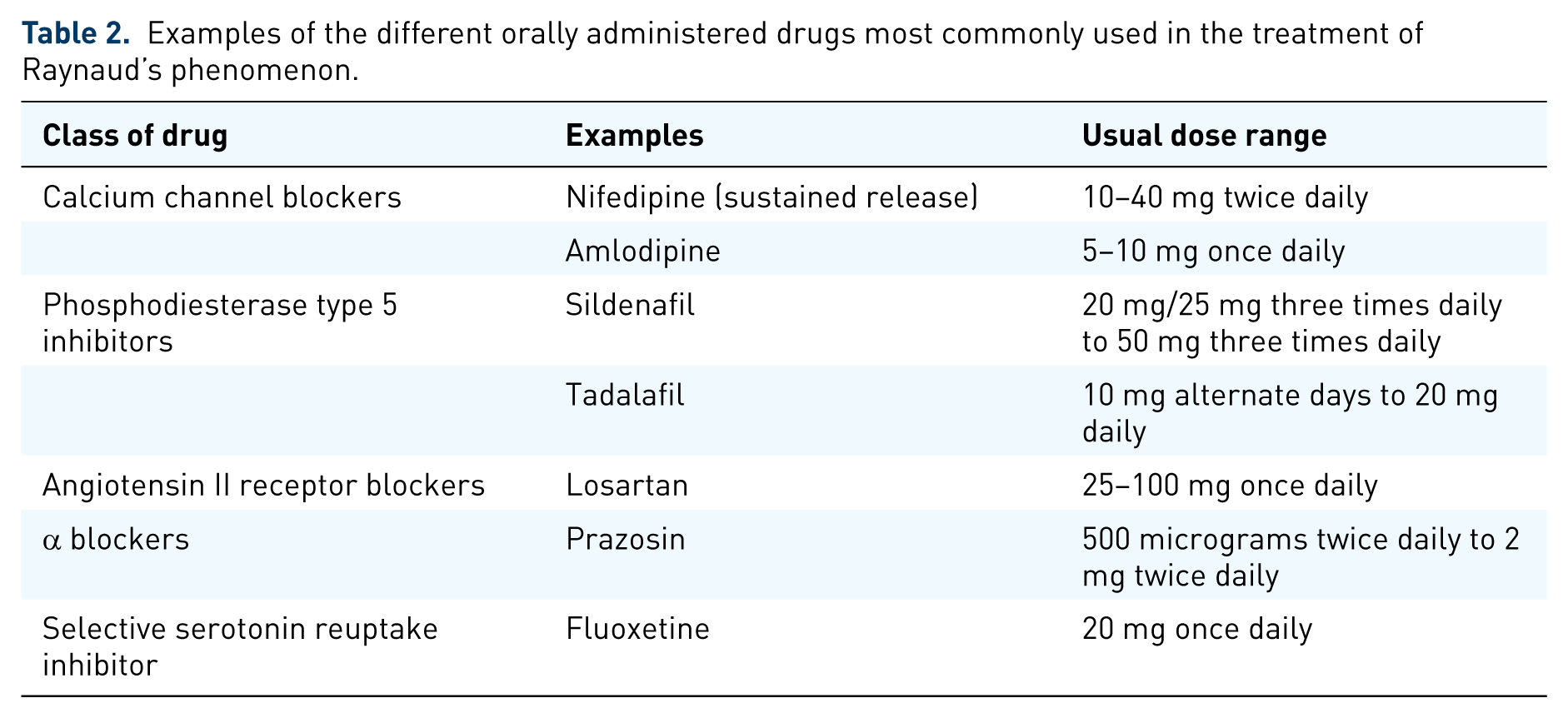
Examples of the different orally administered drugs most commonly used in the treatment of Raynaud’s phenomenon.
Calcium channel blockers
Calcium channel blockers are first-line treatment. However, despite the fact that they are the group of drugs most widely prescribed, there have been relatively few clinical trials examining their efficacy and safety profiles. This is evidenced by a Cochrane review of calcium channel blockers in PRP 31 which included only 296 patients in seven clinical trials. Calcium channel blockers were found to be only minimally effective: there were 1.72 (95% confidence interval 0.60–2.84) fewer RP attacks per week in patients on calcium channel blockers compared with placebo. This was, however, in the context of ‘variable data quality, particularly with regard to outcome measures’ and small sample sizes. 31 Even fewer patients (109 patients) were included in a meta-analysis of calcium channel blockers for SSc-related RP. 32 A disadvantage of calcium channel blockers is that patients frequently report vasodilatory side effects or oedema. A favoured approach 1 is to commence a sustained-release preparation in low dose, then gradually increase the dose as tolerated.
PDE5 inhibitors
PDE5 inhibitors increase the availability or effect of nitric oxide (a potent vasodilator) by inhibiting degradation of cyclic guanosine monophosphate (cGMP): nitric oxide causes smooth muscle relaxation by stimulating soluble guanylate cyclase and increased cGMP. PDE5 inhibitors are being increasingly advocated by rheumatologists for SSc-related RP (the form of RP in which they have been most extensively studied), with many clinicians now using a PDE5 inhibitor as a second choice after (or in addition to) a calcium channel blocker in patients with SSc-related digital vasculopathy. For the practicing rheumatologist, PDE5 inhibitors are therefore probably the most important recent advance in the treatment of ‘uncomplicated’ RP.
What is the evidence base? PDE5 inhibitors have been shown to confer benefit in a number of RCTs,33–36 although it should be noted that these trials were all short term, with a treatment period of 6 weeks or less. A meta-analysis 37 concluded that PDE5 inhibitors have ‘significant but moderate efficacy in secondary RP’. This meta-analysis included six RCTs in 244 patients: three RCTs of tadalafil, two of sildenafil (one modified release) and one of vardenafil. Only 8 of the 244 patients had PRP; all others had RP secondary to connective tissue disease, usually SSc. PDE5 inhibitors conferred benefit in terms of the mean Raynaud’s Condition Score which decreased by −0.46 (95% confidence interval −0.74 to −0.17) (p = 0.002), the daily frequency of RP attacks which decreased by −0.49 (−0.71 to −0.28) (p < 0.0001) and the daily duration of RP attacks which decreased by −14.62 min (−20.25 to −9.00). 37 A recent RCT which was not included in the meta-analysis compared udenafil 100 mg/day to amlodipine 10 mg/day 36 : both had similar efficacy in terms of reducing the frequency of RP attacks. The cost of PDE5 inhibitors has fallen, with patent expiry, and it is likely that these will be increasingly prescribed for RP. There is a need for RCTs examining PDE5 inhibitors in patients with PRP.
Other oral therapies
The evidence base for other oral therapies for RP is very weak,38–41 other drugs sometimes prescribed include angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor antagonists, α blockers, nitrates, and the selective serotonin receptor uptake inhibitor fluoxetine. Some clinicians use an angiotensin II receptor antagonist as first-line treatment, although there has been only one controlled trial (which was open label), which compared losartan 50 mg/day to nifedipine 40 mg/day in patients with PRP and SSc-related RP 42 : 12 weeks’ treatment with losartan conferred benefit in terms of frequency and severity of RP attacks (more so in patients with PRP). Fluoxetine 20 mg daily, administered for 6 weeks, was compared with nifedipine 40 mg daily in an open-label crossover study including patients with primary and secondary RP 43 : frequency and severity of attacks fell on fluoxetine and the authors concluded that larger and placebo-controlled trials were indicated. Fluoxetine has the advantage of not being associated with same vasodilatory side effects as the other drugs mentioned above and may therefore be beneficial in patients intolerant to other therapies. The recent EULAR recommendations 44 for the management of SSc state that fluoxetine ‘might be considered in treatment of SSc-RP attacks.’
Topical vasodilators
Glyceryl trinitrate (GTN) patches can be prescribed for their systemic vasodilatory effects, but are often poorly tolerated. 45 RP is a problem of the extremities and it is therefore disappointing that there are no topical vasodilators marketed for RP for their local effects, the rationale being that these could increase blood flow in the digits without systemic adverse effects. A multicentre, placebo-controlled trial demonstrated benefit from a novel formulation of GTN, MQX-503, 46 in terms of improvement in Raynaud’s Condition Score. The trial included patients with PRP (n = 69) and secondary RP (n = 150, of whom 131 had SSc). MQX-503 gel was applied to the fingers immediately before or within 5 min of onset of a Raynaud’s attack over a 4-week period. 46 Further research into different topical vasodilator therapies for RP is overdue.
Intravenous prostanoid therapy
Although intravenous prostanoids (discussed in the next section) are sometimes used in patients with severe uncomplicated RP, especially in patients with SSc, 44 these are generally reserved for patients whose condition has progressed to digital ulceration or critical ischaemia, or in those who have a history of previous such episodes.
Other therapies
The pathway in Figure 3 includes reference to antiplatelet agents and to statins. There is no good evidence base for either of these approaches. However, there is good evidence for increased platelet activation in patients with SSc, 47 and therefore some clinicians prescribe an antiplatelet agent, 1 especially in patients with SSc and severe RP, in the expectation that this approach may increase microcirculatory flow.
Treatment of RP which has progressed to digital ulceration or critical ischaemia
RP only progresses to tissue damage when secondary to an underlying cause, which for the rheumatologists usually means a connective tissue disease. Because SSc is the connective tissue disease with which RP is most strongly associated (over 95% of patients with SSc have RP), 48 and because SSc-related RP tends to be particularly severe and challenging to manage, most research into connective tissue disease associated digital ulceration and critical ischaemia relates to SSc. Therefore this section (apart from the last subsection ‘Treatment of “inflammatory” vasculopathy’) focuses on SSc, although the basic principles of management are similar across different diseases. These principles of management are very similar for both digital ulceration and critical ischaemia, and so will be considered together. It must be emphasized that although digital ulceration may be a medical emergency requiring immediate hospitalization, critical ischaemia, an example of which is shown in Figure 4, always requires emergency admission to hospital (failure to act quickly risks losing the digit). A number of recent review articles give detailed descriptions of management of SSc-related digital ulcers.4,49,50 The UK Scleroderma Study Group consensus best practice pathways include flow charts outlining the approach to treatment 12 : Figure 5 gives an updated pathway for digital ulceration. As with the RP flowchart (Figure 3), PDE5 inhibitors are now positioned ‘higher up’.
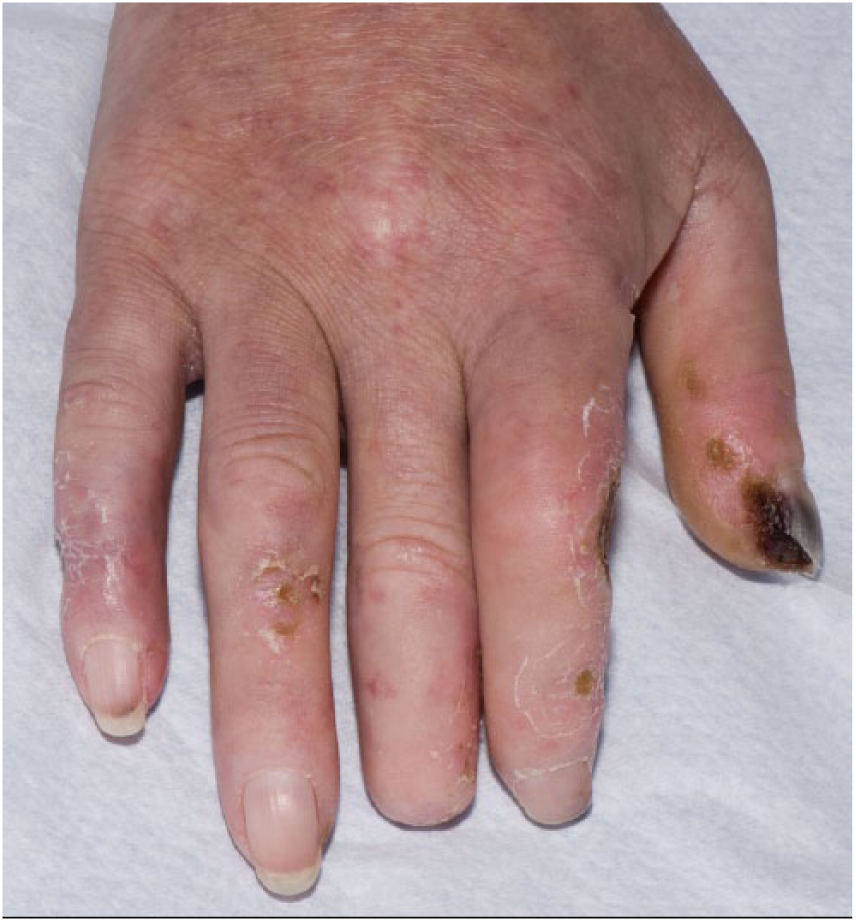
Critical digital ischaemia of the thumb in a patient with systemic sclerosis and very severe digital vasculopathy (digital ulcers are also present).

Establishing the diagnosis early and removing any identifiable cause
Patients with SSc, or with another connective tissue disease and severe RP, should be educated to seek medical advice urgently in the event of a digital ulcer or a permanent colour change developing in one or more fingers. Ideally an ‘open-door’ policy should be in place to allow rapid assessment and treatment. The concern is that without early active intervention, an ulcer may become infected (infection could then spread to bone), or a critically ischaemic digit may become unviable.
Even in a patient with known connective tissue disease, always consider the possibility of another underlying or contributory cause, 51 especially proximal (large) vessel disease, a coagulopathy or a concomitant vasculitis, all of which would require specific treatment.
Analgesia and antibiotics
Digital ulcers and critical ischaemia can be excruciatingly painful, often keeping the patient awake at night. Adequate analgesia is an essential part of management and is now being recognized.52,53 Opioids may be required in the short term. Antibiotics should be given if there is suspected or definite infection.
Vasoactive therapies
The past 10 years have seen major advances in the therapeutic armamentorium for SSc-related digital ulcers and these are incorporated into the National Health Service (NHS) England current clinical commissioning policy 54 for treatment of SSc-related digital ulceration and also into the British Society for Rheumatology and British Health Professionals in Rheumatology guideline for treatment of SSc. 55 In summary, the NHS England policy 54 recommends the following steps (if a step is ineffective, then the next step should be proceeded to):
Standard medical therapy (e.g. calcium channel blockers, ACE inhibitors, losartan, fluoxetine).
Sildenafil 25 mg three times daily, increasing to 50 mg three times daily if necessary.
Intravenous prostanoid (usually iloprost) up to a frequency of every 6–8 weeks if necessary.
Intravenous prostanoid and sildenafil in combination.
Bosentan.
The main advances in the last 10 years are first the licencing of bosentan, a dual endothelin (ET)-1 receptor antagonist (antagonizing ETA and ETB ET-1 receptors), for prevention of recurrent digital ulcers in patients with SSc; and second, the increased use of PDE5 inhibitors.
What is the evidence to underpin the use of PDE5 inhibitors, intravenous prostanoids and bosentan?
PDE5 inhibitors
The SEDUCE study (of 83 evaluable patients with SSc, with 192 digital ulcers), compared 12 weeks of treatment with sildenafil 20 mg three times daily with placebo). 56 Sildenafil conferred some benefit with a greater healing rate in the sildenafil group compared with placebo at week 8 (p = 0.01) and week 12 (p = 0.03), although the primary endpoint (time to healing) was not reached.
Intravenous prostanoid therapy
Intravenous therapy with prostacyclin analogues is well established in the treatment of patients in whom RP is very severe and has progressed to digital ulceration. Most experience is with iloprost but epoprostenol 57 may also be used. Intravenous prostanoids reduce frequency and severity of RP attacks and heal digital ulcers.58,59 However, they require hospitalization, which is expensive and inconvenient for patients, and are often associated with systemic vasodilator side effects.
To date, trials of oral prostanoids have been disappointing. However, supplementing the prostacyclin pathway with oral therapies has recently been revisited. A multicentre RCT of oral treprostinil in 147 patients with SSc-related digital ulcers 60 showed a small (statistically insignificant) reduction in net ulcer burden (–0.43 ulcers) compared with placebo (–0.10 ulcers) after 20 weeks of treatment and patients receiving treprostinil in an open-label extension also demonstrated a small reduction in net ulcer burden. A subsequent retrospective study of 51 patients in whom treprostinil was withdrawn after the open-label extension and for whom follow-up data were available 61 found that digital ulcer burden significantly increased at 3–6 months and at 6–12 months after discontinuing treprostinil, suggesting that despite the failure of the RCT 60 to meet its primary endpoint (change in net digital ulcer burden), further study of oral treprostinil is warranted. In a recent RCT including 74 patients with SSc-related RP, 62 there was no reduction in the number of RP attacks in patients receiving selexipag, an oral IP prostacyclin receptor agonist, compared with placebo. At present, treatments aimed at supplementing the prostacyclin pathway are confined (in most countries including the UK) to the intravenous route.
ET-1 receptor antagonists
Bosentan is licenced for the prevention of recurrent digital ulcers in patients with SSc. It has been shown in two multicentre RCTs,63,64 comparing bosentan with placebo, to reduce the number of new ulcers, although there was no effect on the healing of existing ulcers. Macitentan, another dual ET-receptor antagonist (but with sustained receptor binding and increased tissue penetration compared with bosentan) has recently been trialled in SSc-related digital ulceration. Disappointingly the DUAL-1 and DUAL-2 studies 65 which randomized 289 and 265 patients respectively did not show benefit from 16 weeks of treatment with macitentan 3 mg daily or 10 mg daily compared with placebo in the cumulative number of new ulcers (the primary endpoint). DUAL-2 was halted prematurely on the recommendation of the independent data monitoring committee. There have been no major studies of any other ET-1 antagonists in SSc-related digital vasculopathy.
Other drug treatments with effects on the vasculature
The rationale for antiplatelet agents was discussed earlier. Is has been suggested that statins confer benefit in SSc-related digital ulceration66,67 but further studies are required. In patients with critical ischaemia, especially if progressive, then short-term anticoagulation could be considered but it must be stressed that there is no good evidence base for this approach. 12
Procedural therapies, including surgery
Botulinum toxin injections
These have attracted increasing interest in recent years, 68 recent studies including a prospective study of 20 patients with SSc reporting improvement in hand function after 8 weeks, 69 and a case series of 10 patients with SSc (5 had digital ulcers) reporting benefit in terms of RP, pain, skin temperature recovery after cold water immersion and digital ulcer healing. 70 Results of RCTs are eagerly awaited.
Fat grafting
Autologous fat grafting or injection of adipose tissue derived stromal or stem cells71,72 is now attracting substantial interest in SSc-related digital vasculopathy and at least one controlled trial is underway. Although the exact mechanism of action of fat grafting in this clinical context is not fully understood, it is likely that the transplanted cells have proangiogenic, antifibrotic, anti-inflammatory and immunomodulatory effects.71,72
Surgery
A number of different surgical procedures have been advocated for severe SSc-related digital vasculopathy, including surgical debridement, digital sympathectomy and amputation. 73 Digital sympathectomy73,74 is probably being performed increasingly in specialist centres and a number of case series and observational studies have now been reported. A recent retrospective study of 17 patients with SSc (26 hands operated on) 75 reported symptomatic improvement in pain in 92.3% of hands and ulcer healing in all patients. A recent survey of 500 rheumatologists in the United States, of whom 107 responded, highlighted the importance of multidisciplinary team working between hand surgeons and rheumatologists. 76 A systematic review concluded that the evidence base for surgical procedures (including sympathectomy) for RP is lacking 77 ; however, this is perhaps unsurprising given the relatively small numbers of patients coming to surgery and the difficulties in mounting clinical trials.
Treatment of ‘inflammatory’ vasculopathy
In patients with SSc, the vascular abnormalities which drive digital ischaemia are primarily noninflammatory and are a result of structural and functional disease of the microcirculation and of the digital artery with obliterative intimal thickening of the small arteries: any inflammation is mild 78 and great caution is required before considering steroid therapy. 79 However, in a patient with a primarily inflammatory connective tissue disease, for example systemic lupus erythematosus, or with overt vasculitis, the situation is different and corticosteroids or immunosuppressants might then be indicated. Each case must be considered individually and the risks and benefits of corticosteroids and immunosuppressants carefully considered.
Possible future therapies
In addition to fat grafting for SSc-related digital vasculopathy, already mentioned above because it is used in certain centres but is still very much ‘emerging’, other new treatments are on the horizon or deserve investigation. These include topical therapies, applied to the digits to improve blood flow locally without systemic (including adverse) effects. There are different ways of increasing blood flow locally, including direct application of (for example) GTN to digital ulcers 80 and iontophoresis.81,82
New therapeutic strategies are badly needed: a recent survey indicated that only 16% of 443 patients with RP felt that current medication was effective. 83 It is therefore encouraging that large multicentre studies of RP and digital ulceration are now being mounted, at least in patients with SSc. With increasing interest in developing reliable outcome measures for RP, RCTs in PRP should also increase.
Conclusion
RP provides a window of opportunity to the rheumatologist to make an early diagnosis of an underlying connective tissue disease. Treatment of RP in patients with rheumatological disease can be challenging, especially when it has progressed to digital ulceration or critical ischaemia. Educating patients to seek urgent medical advice if they develop an ulcer or permanent discolouration (suggesting critical ischaemia) is a key aspect of management. While calcium channel blockers remain first-line treatment, recent advances in medical therapy include PDE5 inhibitors (for RP and for digital ulceration, particularly for patients with SSc) and bosentan for patients with SSc and recurrent ulcers. A number of procedural therapies (botulinum toxin injections, fat grafting and digital sympathectomy) are attracting increasing attention, although controlled trials and longer-term observational studies are required to establish an evidence base for their exact role. Future therapies, in development, should include topical treatments applied locally to the digits and free of systemic adverse effects. Although the evidence base for many of the treatments used for RP remains weak, it is encouraging that the last 10 years have seen a number of multinational RCTs examining different treatments for RP and SSc-related digital ulceration.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
ALH has done consultancy work for Actelion, served on a Data Safety Monitoring Board for Apricus, received research funding and speaker’s fees from Actelion, and speaker’s fees from GSK.