
Abstract
Objective:
To evaluate the safety, tolerability, and efficacy of adding milnacipran to pregabalin in patients with fibromyalgia who have experienced an incomplete response to pregabalin.
Methods:
In this randomized, multicenter, open-label study, patients received pregabalin 300 or 450 mg/day during a 4- to 12-week run-in period. Patients with weekly recall visual analog scale (VAS) pain score of at least 40 and up to 90, Patient Global Impression of Severity score of at least 4, and Patient Global Impression of Change (PGIC) score of at least 3 were classified as incomplete responders and randomized to continue pregabalin alone (n = 180) or receive milnacipran 100 mg/day added to pregabalin (n = 184). The primary efficacy parameter was responder status based on PGIC score of up to 2. The secondary efficacy parameter was change from randomization in weekly recall VAS pain score. Safety parameters included adverse events (AEs), vital signs, and clinical laboratory tests.
Results:
The percentage of PGIC responders was significantly higher with milnacipran added to pregabalin (46.4%) than with pregabalin alone (20.8%; p < 0.001). Mean improvement from randomization in weekly recall VAS pain scores was greater in patients receiving milnacipran added to pregabalin (−20.77) than in patients receiving pregabalin alone (−6.43; p < 0.001). During the run-in period, the most common treatment-emergent AEs with pregabalin were dizziness (22.8%), somnolence (17.3%), and fatigue (9.1%). During the randomized period, the most common treatment-emergent AEs with milnacipran added to pregabalin were nausea (12.5%), fatigue (10.3%), and constipation (9.8%).
Conclusions:
In this exploratory, open-label study, adding milnacipran to pregabalin improved global status, pain, and other symptoms in patients with fibromyalgia with an incomplete response to pregabalin treatment.
Introduction
Fibromyalgia is a complex pain disorder that affects 2–4% of the US population, more commonly in women than in men [Wolfe et al. 1995]. In addition to chronic widespread pain, patients with fibromyalgia often report other potentially debilitating symptoms such as fatigue, sleep disturbances, cognitive problems, impaired physical function, and depressed mood [Bennett, 2009; Arnold et al. 2011]. A number of comorbid disorders are also associated with fibromyalgia, including other chronic pain conditions and psychiatric illnesses [Arnold, 2008; Lachaine et al. 2010].
Over the past decade, a number of studies have been conducted to evaluate the safety and efficacy of various medications in patients with fibromyalgia. Based on results from large, randomized, placebo-controlled studies, pregabalin (PGN), duloxetine, and milnacipran (MLN) have been approved in the United States for the management of fibromyalgia [Pfizer Inc., 2012; Eli Lilly and Company, 2012; Forest Laboratories, Inc., 2012]. Although clinical trials with the approved medications have demonstrated improvements in pain and other symptoms of fibromyalgia, none of these medications are effective in all patients. Due to the multisymptomatic nature of fibromyalgia [Arnold, 2009], some patients may require the addition of another medication in order to achieve satisfactory clinical improvements [McCarberg, 2012].
Based on what is known about the pathophysiology of fibromyalgia and the mechanisms of action of approved medications, adding MLN to PGN may be a rational treatment strategy for some patients with fibromyalgia [Mease and Seymour, 2008]. MLN is a dual reuptake inhibitor whose mechanism of analgesic efficacy is postulated to be that of increasing norepinephrine and serotonin signaling in the descending pain pathways, which results in inhibition or modulation of pain signaling in the central nervous system [Kranzler and Gendreau, 2010]. PGN, a ligand for the α2-δ subunit of voltage-gated calcium channels, is thought to reduce the transmission of nociceptive signals via ascending pain pathways by modulating the calcium-dependent release of excitatory neurotransmitters such as glutamate and substance P [Arnold et al. 2010b]. Studies in animal models suggest that PGN may also affect norepinephrine levels in the descending pathways [Takeuchi et al. 2007]. In addition to having potentially complementary mechanisms of action in the treatment of chronic central pain, both MLN and PGN have a low potential for pharmacokinetic drug–drug interactions; however, pharmacodynamic interactions can occur with each of these drugs [Forest Laboratories, Inc., 2012; Pfizer Inc., 2012].
Several studies have examined the effects of add-on or combination therapies in patients with fibromyalgia [Goldenberg et al. 1996; Bennett et al. 2003; Calandre et al. 2007, 2011]. However, these studies included medications not approved for the management of fibromyalgia; in addition, some studies included small patient populations (n < 20) or were uncontrolled. Nonetheless, combination therapies may be clinically beneficial, as suggested by the results of a recently published study that examined changes in prescribing patterns of medications commonly used to treat fibromyalgia, along with changes in clinical domains relevant in fibromyalgia. The study, which included data from 232 patients from 15 rheumatology clinics, found that the combination of anticonvulsants and antidepressants was more beneficial than treatment with either drug class alone [Rivera et al. 2012]. The study summarized in the present report evaluated a combination of US Food and Drug Administration approved medications from these two different classes, and represents one possible treatment strategy for managing patients with fibromyalgia. The aim of this randomized, open-label study was to evaluate the efficacy, safety, and tolerability of adding MLN to PGN in patients with fibromyalgia who had an incomplete response to PGN. It is expected that findings from this study, which is the first to evaluate the combined use of two approved medications with complementary mechanisms of action, will offer preliminary evidence for the efficacy and safety of combining these medications in patients with fibromyalgia.
Methods
Study overview
A multicenter, randomized, open-label study was conducted in the United States (60 sites) to evaluate the efficacy and tolerability of MLN when added to PGN in patients with an incomplete response to PGN for the treatment of fibromyalgia. The study was conducted from November 2008 to January 2010 [ClinicalTrials.gov identifier: NCT00797797]. The study included a 4- to 12-week, open-label, PGN run-in period, followed by an 11-week period in which eligible patients (i.e. incomplete responders) were randomized 1:1 to continue PGN alone or to receive MLN + PGN. The study protocol and informed consent documents were reviewed and approved by the institutional review boards or independent ethics committees at each of the participating study centers. Written informed consent was obtained from all participants.
Patients
Eligible participants were outpatients aged 18–70 years who met the 1990 American College of Rheumatology criteria for fibromyalgia [Wolfe et al. 1990]. At screening, patients were required to have a mean weekly recall visual analog scale (VAS) pain score of at least 40 (range 0–100, with 100 indicating worst possible pain). Patients taking concomitant medications that could influence pain perception and who had a VAS pain score less than 40 at screening were allowed a 1- to 4-week washout period and an opportunity to repeat the VAS pain assessment after discontinuing prohibited medications.
Patients with major medical disorders or uncontrolled medical conditions were excluded from the study, including those with recent myocardial infarction or stroke, active liver disease, documented autoimmune disease, active cancer, significant gastrointestinal disorders, pulmonary dysfunction, severe chronic obstructive pulmonary disease, unstable diabetes, or unstable thyroid disease. Patients with any of the following criteria were also excluded from the study: exposure to MLN, duloxetine, or any investigational drug within 30 days of screening; significant risk of suicide or severe psychiatric illness based on investigator’s judgment or patient responses on the Beck Depression Inventory [Beck et al. 1961]; history of behavior that could potentially prohibit compliance for the duration of the study; substance abuse; pregnancy or breastfeeding; unacceptable contraception; prostate enlargement or other genitourinary disorder; renal impairment (creatinine clearance < 60 ml/min); uncontrolled narrow-angle glaucoma; body mass index of at least 40. Based on investigator judgment, patients with any medical condition that might interfere with study participation, confound the interpretation of study results, or endanger the patient were also excluded from the study.
Medications that act on the central nervous system and are commonly used to treat fibromyalgia symptoms were prohibited, including antidepressants, antiepileptic agents, opiates and related analgesics (e.g. oxycodone, codeine, tramadol, narcotic patches), dopamine agonists, and sodium oxybate. Other excluded drugs included stimulants used for attention deficit hyperactivity disorder, anorectic agents, and buspirone. Patients requiring washout of excluded medications were allowed to do so during the PGN run-in treatment. Medication washout periods were 1–4 weeks, depending on the type of medication requiring washout. Use of acetaminophen, aspirin, and nonsteroidal anti-inflammatory agents was permitted. In addition, patients requiring short-term pain rescue medication were allowed opioid analgesics; however, opioids were not permitted within 7 days of scheduled study visits. Triptans were permitted for acute migraine treatment. Nonbenzodiazepine hypnotics were allowed for patients requiring treatment of insomnia.
Open-label run-in procedures
At screening (visit 1), eligible participants were enrolled in the 4- to 12-week open-label run-in period to evaluate their response to PGN monotherapy. PGN doses were administered according to the treatment recommendations for fibromyalgia in the package insert [Pfizer Inc., 2012]. Most patients (85%) were not currently taking PGN at enrollment. These participants initially received PGN 75 mg twice daily (150 mg/day), increased to 150 mg twice daily (300 mg/day) within 1 week based on efficacy and tolerability, followed by an increase to 225 mg twice daily (450 mg/day) if tolerated. Dosages for patients who entered the study while taking PGN were adjusted to 300 or 450 mg/day. For patients entering the study receiving less than 450 mg/day, PGN dosage was escalated to 450 mg/day if tolerated; for those receiving more than 450 mg/day, dosage was lowered to 450 mg/day. Patients unable to tolerate PGN 300 mg/day were not eligible for randomization.
Study visits occurred every 4 weeks during the open-label treatment as needed. To qualify for randomization after the first 4 weeks of open-label treatment, patients had to complete at least 2 weeks of stable dosing with PGN 300 or 450 mg/day and be classified as an incomplete responder. PGN incomplete responders were defined as patients who met all three of the following criteria: weekly recall VAS pain score of at least 40 and up to 90; Patient Global Impression of Severity (PGI-S) score of 4 or higher (‘moderately ill’ to ‘extremely ill’); and a Patient Global Impression of Change (PGIC) score of 3 (‘minimally improved’) or higher (‘no change’ to ‘very much worse’). Patients not meeting all of the incomplete responder criteria continued receiving open-label PGN treatment for an additional 4 or 8 weeks. After a total of 12 weeks of PGN treatment, patients not meeting the incomplete responder criteria were discontinued from the study.
Randomized, open-label treatment period procedures
After the PGN run-in phase, eligible patients were randomized (1:1) to continue PGN treatment alone or to receive MLN 100 mg/day added to their current PGN regimen for an additional 11 weeks. Patients randomized to receive MLN + PGN underwent a 1-week dose escalation to MLN 100 mg/day followed by 10 weeks of stable dose treatment.
Outcome measures and analyses
The primary efficacy parameter was PGIC responder status at end of the study, defined as the proportion of patients who rated their overall improvement since screening as ‘very much improved’ (score = 1) or ‘much improved’ (score = 2) based on a seven-point scale ranging from 1 to 7 (‘very much worse’). PGIC was assessed at randomization and at each study visit (weeks 2, 6, and 11). Patient-reported global status has been identified by the Outcome Measures in Rheumatology Fibromyalgia Working Group as a core outcome to be evaluated in clinical studies [Mease et al. 2009a], and the definition of PGIC responder status used in this study was consistent with recommendations from the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials [Dworkin et al. 2005]. In addition, since two different classes of drugs were evaluated in this open-label study, the PGIC was chosen as a measure that would evaluate multiple domains (e.g. pain relief, improved functioning, improvements in fatigue and cognition, side effects) in all patients.
The secondary efficacy parameter was change from randomization in the weekly recall VAS pain score. A post hoc analysis was also conducted to identify the percentage of patients with clinically meaningful pain improvements, defined as at least 30% decrease from randomization in VAS pain score [Emshoff et al. 2011].
Additional efficacy analyses included changes in physical and mental functioning, as measured by Short Form-36 Health Survey (SF-36) Physical Component Summary (PCS) and Mental Component Summary (MCS) scores [Ware and Sherbourne, 1992]. Changes in fatigue were evaluated using the Multidimensional Fatigue Inventory (MFI) [Smets et al. 1995]; cognitive changes were evaluated using the Multiple Ability Self-Report Questionnaire (MASQ) [Seidenberg et al. 1994]. Secondary and additional efficacy measures were assessed at screening, randomization, and each study visit.
Tolerability and safety assessments
Adverse events (AEs) were recorded at study visits, by telephone at week 1 during the randomized period, and during any communication with a patient or patient representative. Patients were asked a nonleading question (e.g. ‘How do you feel?’) and all patient responses were recorded in individual case report forms, along with the investigator’s assessment of severity, causality, and seriousness. All reported events were coded using Medical Dictionary for Regulatory Activities (MedDRA) terms, version 11.0 or newer. For the PGN run-in period, treatment-emergent AEs were defined as those events that started or worsened between the first dose of run-in treatment and the first dose of randomized treatment. Analyses were also conducted on the subgroups of patients who received PGN prior to entering the run-in period and those who were newly exposed to PGN treatment. For the randomized period, treatment-emergent AEs were defined as those events that started or worsened in severity after the first dose of randomized treatment; AEs were collected for up to 30 days after the last randomized dose.
Vital signs and body weight were assessed at screening, randomization, and each clinic visit (except week 2 of the randomized period for weight). Clinical laboratory tests were conducted at screening, randomization, and at weeks 6 and 11 of the randomized period.
Statistical analysis
Power calculations suggested that approximately 160 patients per randomized treatment group would be needed to provide 80% power for a two-sided test at the 0.05 level of significance, based on an expected 10% difference between treatment groups in PGIC responder rates. For the run-in period, the safety population included all screened patients who received at least one dose of PGN. For the randomized period, the safety population included all randomized patients who received at least one dose of randomized study drug. All patients in the randomized safety population who had at least one post-randomization PGIC assessment were included in the intent-to-treat (ITT) analyses.
For the primary efficacy outcome, the difference between treatment groups was analyzed using a logistic regression model, with treatment group as the only explanatory variable. Missing values were imputed using a modified last observation carried forward (LOCF) approach in which patients discontinuing the study due to lack of efficacy or AEs were defined as nonresponders; missing data due to other reasons were imputed using the LOCF approach. Recent recommendations on data imputation indicate that no single method is adequate [Walton, 2009]. Therefore, several sensitivity analyses were conducted using observed cases, baseline observation carried forward, and LOCF approaches. A generalized linear mixed model for repeated measures based on observed cases was also used to assess robustness of the primary efficacy analysis. Secondary and additional efficacy assessments were analyzed using an analysis of covariance model, with treatment group and study center as factors and the baseline value as a covariate. Baseline was defined as the randomization visit for these statistical analyses. All statistical tests for the randomized treatment period were two-sided hypothesis tests performed at the 0.05 level of significance.
Descriptive statistics were used to analyze safety outcomes. Potentially clinically significant changes in vital signs or laboratory values were analyzed in patients with a nonmissing randomization value and at least one postrandomization assessment.
Results
Patients
A total of 850 patients were screened for eligibility (Figure 1). During the 4- to 12-week PGN run-in period, 705 patients received at least one dose of PGN; 600 of these patients were newly exposed to PGN and 105 entered the study already receiving PGN treatment. At the end of the PGN run-in period, 101 patients were classified as PGN responders and were discontinued from the study.
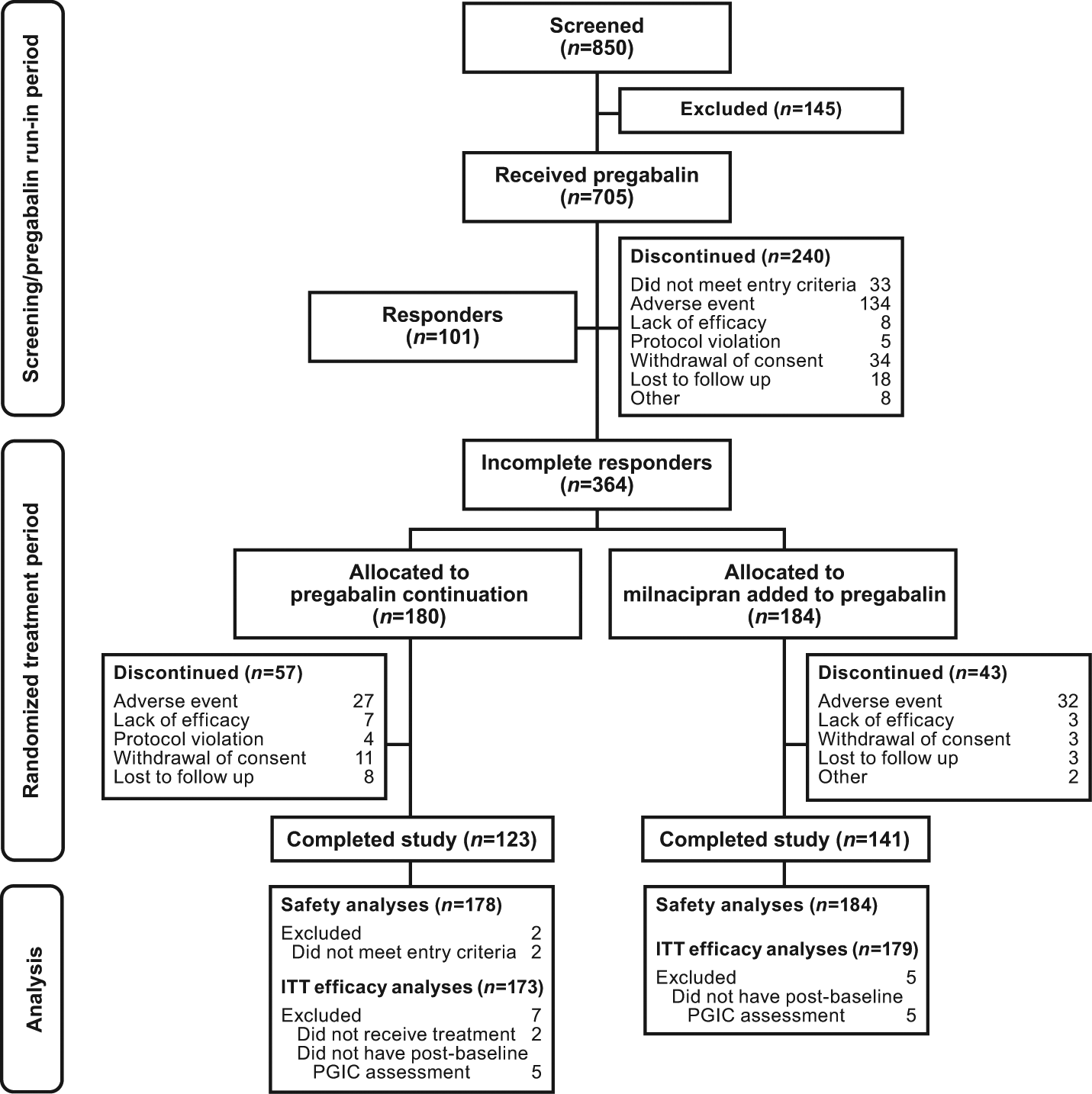
Flow diagram of study progress. ITT, intent to treat; PGIC, Patient Global Impression of Change.
A total of 364 patients (51.6%) completed the PGN run-in period, were classified as incomplete responders, and were randomized to continue PGN alone (n = 180) or receive MLN added to PGN (MLN + PGN, n = 184). Of these randomized patients, 70 had received prior PGN (PGN alone, n = 32; MLN + PGN, n = 38) and 294 received no prior PGN (PGN alone, n = 148; MLN + PGN, n = 146). Two patients in the randomized population did not receive study medication and were excluded from safety and ITT efficacy analyses. Ten patients who did not have at least one post-randomization PGIC assessment were excluded from the ITT efficacy analyses. More patients in the MLN + PGN group (76.6%) completed the study than in the PGN group (68.3%). For both groups, AEs were the most frequently reported reason for discontinuing the study.
Demographics and clinical characteristics were similar between treatment groups (Table 1). Most patients were women (90.6%) and white (90.3%); mean age was 49.4 years. Mean VAS pain scores at randomization ranged from 66.8 to 67.6, indicating moderate to severe pain in this patient population [Bennett et al. 2003]. Patients also had impaired physical and mental function at randomization, as indicated by mean SF-36 PCS and MCS scores [Ware and Sherbourne, 1992].
Demographics and patient characteristics, ITT population.
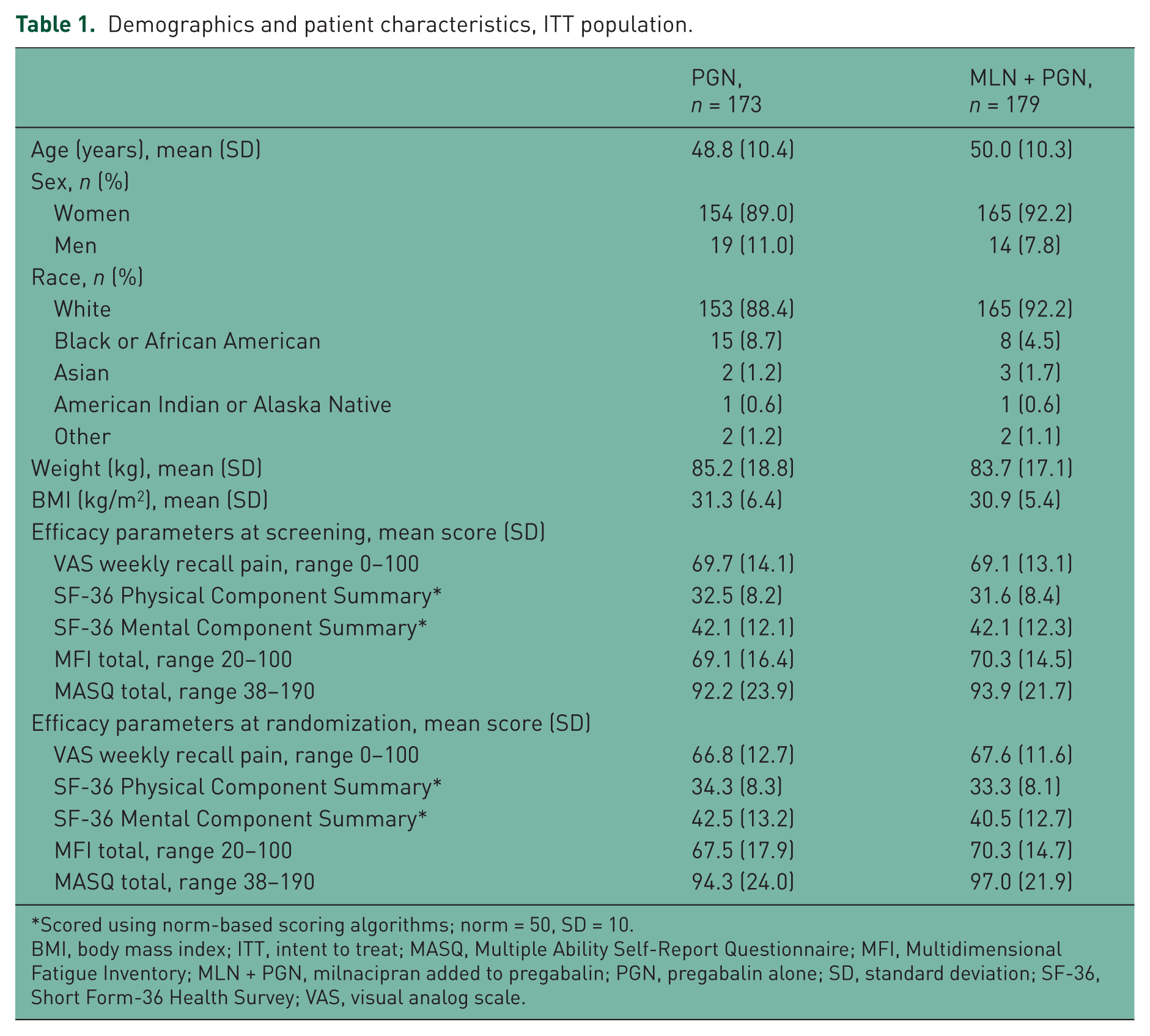
Scored using norm-based scoring algorithms; norm = 50, SD = 10.
BMI, body mass index; ITT, intent to treat; MASQ, Multiple Ability Self-Report Questionnaire; MFI, Multidimensional Fatigue Inventory; MLN + PGN, milnacipran added to pregabalin; PGN, pregabalin alone; SD, standard deviation; SF-36, Short Form-36 Health Survey; VAS, visual analog scale.
During the PGN run-in period in patients subsequently randomized as incomplete responders, mean improvements from screening [± standard error of the mean (SEM)] were as follows: VAS pain, -2.2 (± 0.8); SF-36 PCS, 1.6 (± 0.3); and MFI total, -0.7 (± 0.6). On the PGIC scale, 171 (47.0%) randomized patients reported no change or worsening and 193 (53.0%) reported minimal improvements from screening during the PGN run-in period. A worsening from screening in MASQ total score (2.4 ± 0.7) was also found. Of the 364 randomized patients, 83 (22.8%) received PGN 300 mg/day and 281 (77.2%) received PGN 450 mg/day.
Efficacy
At the end of the randomized, open-label treatment period, the proportion of PGIC responders (‘much improved’ or ‘very much improved’) was significantly higher in patients receiving MLN + PGN (46.4%) than in patients continuing PGN alone (20.8%; p < 0.001) (Figure 2). Similar findings were observed on all sensitivity analyses, with approximately twice as many patients receiving MLN + PGN classified as PGIC responders: MLN + PGN, 45.8–59.2%; PGN, 19.7–27.6%; all p < 0.001.

PGIC responder rates, defined as the percentage of patients rating their overall improvement since entering the study as very much or much improved (PGIC score ≤ 2). BOCF, baseline observation carried forward; GLMM, generalized linear mixed model; LOCF, last observation carried forward; MLN + PGN, milnacipran added to pregabalin; mLOCF, modified last observation carried forward; n, number of responders; N, number of patients; OC, observed case; PGIC, Patient Global Impression of Change; PGN, pregabalin alone. *p < 0.001 versus PGN. $Percentages are estimates of treatment group response probabilities from the GLMM.
Patients receiving MLN + PGN had significantly greater mean improvement from randomization in weekly recall VAS pain score than patients continuing PGN alone (Figure 3). Least squares mean changes (± SEM) from randomization to end of study were as follows: MLN + PGN, -20.77 (± 1.92); PGN, −6.43 (± 1.93); p < 0.001 (LOCF). Significant differences between treatment groups were observed within 2 weeks following the addition of MLN to PGN and were maintained at all subsequent study visits (all visits, p < 0.001 versus PGN). In addition, the percentage of patients with at least 30% pain improvement was higher in the group that received MLN + PGN than in the group that continued PGN alone: MLN + PGN, 45.8%; PGN, 19.7% (LOCF).

Least squares mean change from randomization in weekly recall VAS pain score (LOCF). LOCF, last observation carried forward; MLN + PGN, milnacipran added to pregabalin; PGN, pregabalin alone; VAS, visual analog scale. *p < 0.001 versus PGN.
Patients receiving MLN + PGN also demonstrated significantly greater mean improvements from randomization in physical functioning (SF-36 PCS), mental functioning (SF-36 MCS), fatigue (MFI total), and self-reported cognition (MASQ total) than patients continuing PGN alone (Table 2). In addition, significant improvements with MLN + PGN were found in all SF-36 PCS subscales (p < 0.001 versus PGN), all MFI domains (p < 0.01 versus PGN), and all MASQ domains (p < 0.05 versus PGN).
Least squares mean changes from randomization to last study visit in additional efficacy outcomes (LOCF).

ANCOVA, analysis of covariance; CI, confidence interval; LOCF, last observation carried forward; LS, least squares; MASQ, Multiple Ability Self-Report Questionnaire; MFI, Multidimensional Fatigue Inventory; MLN + PGN, milnacipran added to pregabalin; PGN, pregabalin alone; SE, standard error; SF-36, Short Form-36 Health Survey.
Safety
Treatment-emergent AEs for the randomized and run-in periods are shown in Table 3. The most common treatment-emergent AEs during the PGN run-in period were dizziness (22.8%), somnolence (17.3%), fatigue (9.1%), peripheral edema (8.9%), and weight increased (8.1%). The types of treatment-emergent AEs were similar to what has been reported in other PGN studies [Pfizer Inc., 2012].
Treatment-emergent adverse events reported in at least 5% of total patients during the run-in period or in at least 5% of patients in either treatment group during the randomized period.

Treatment-emergent adverse events defined as adverse events that occurred with or after the first dose of pregabalin treatment but prior to the first dose of randomized study medication (for randomized patients) or up to 30 days after last pregabalin dose (for nonrandomized patients).
Treatment-emergent adverse events defined as adverse events that were newly reported or increased in severity during the randomized period.
MLN + PGN, milnacipran added to pregabalin; new PGN, patients newly exposed to pregabalin treatment during the run-in period; PGN, pregabalin alone; prior PGN, patients receiving pregabalin prior to entering the study.
A total of 114 (64.0%) patients continuing PGN alone and 141 (76.6%) patients receiving MLN + PGN experienced at least one treatment-emergent AE that was not present before randomization or that worsened during the randomized, open-label period. Most (94%) treatment-emergent AEs during the randomized period were mild or moderate in severity. Peripheral edema (8.4%) and weight increase (8.4%) were the most commonly reported treatment-emergent AEs in patients who continued receiving PGN alone (Table 3). Consistent with other MLN studies [Forest Laboratories, Inc., 2012], nausea was the most common treatment-emergent AE in patients who received MLN + PGN in this study (12.5%), although the incidence was lower than has been previously reported. Fatigue (10.3%), constipation (9.8%), and headache (9.2%) were the other most common treatment-emergent AEs in these patients.
During the randomized period, 26 (14.6%) patients continuing PGN alone and 32 (17.4%) patients receiving MLN + PGN discontinued due to an AE; AEs leading to discontinuation in at least 1% of patients in either treatment group are listed in Table 4. Serious AEs were reported in six (3.4%) patients who continued PGN and in five (2.7%) patients who received MLN + PGN. The two serious AEs judged by investigators as possibly related to study medication were irritable bowel syndrome and hypertensive urgency, each of which occurred in one patient in the MLN + PGN group. Both patients discontinued the study.
Discontinuations due to adverse events reported in at least 1% of patients in either treatment group during the randomized period, based on the safety population.

Nonrandomized patients who discontinued during the run-in period due to adverse events.
Patients who discontinued during the randomized period due to adverse event; for some of these patients adverse events began during the run-in period.
MLN + PGN, milnacipran added to pregabalin; new PGN, patients newly exposed to pregabalin treatment during the run-in period; PGN, pregabalin alone; prior PGN, patients receiving pregabalin prior to entering the study.
Mean changes in sitting blood pressure and heart rate were analyzed for the PGN run-in period, randomized open-label treatment period, and overall study (Table 5). Mean decreases or minimal changes in these vital signs were found during the PGN run-in period. During the randomized period, mean increases in blood pressure and heart rate were found in patients who received MLN + PGN. However, when analyses for this group were conducted to include changes that had occurred during the PGN run-in period (i.e. mean changes from screening to end of study), mean increases in blood pressure and heart rate were smaller than the increases observed during the randomized period.
Mean changes in blood pressure and heart rate.

From screening to end of pregabalin run-in period, defined as the last available assessment in the run-in period.
From randomization to end of study, defined as the last available assessment in the randomized, open-label treatment period.
Only includes patients with a reference value (screening or randomization) and at least one postrandomization value.
BP, blood pressure; MLN + PGN, milnacipran added to pregabalin; PGN, pregabalin alone; SD, standard deviation.
During the randomized period, potential clinically significant changes in vital signs were found in one patient receiving MLN + PGN (heart rate ≥ 120 bpm and increase from randomization ≥ 20 bpm) and three patients receiving PGN (one with diastolic blood pressure ≥ 110 mmHg and an increase from randomization ≥ 10 mmHg; two with heart rate ≤ 50 bpm and a decrease from randomization ≥ 15 bpm).
MLN + PGN did not increase body weight. Mean changes from randomization to end of study were −0.04 kg for patients receiving MLN + PGN and +0.4 kg for patients continuing PGN alone. Potentially clinically significant weight gain (≥ 7% increase from randomization) was found in seven patients receiving PGN alone and three patients on MLN + PGN. Two patients in each treatment group had potentially clinically significant weight loss (≥ 7% decrease from randomization).
Mild elevations in alanine aminotransferase (ALT) and aminotransferase levels (AST) have been found in some patients with fibromyalgia treated with MLN [Forest Laboratories, Inc., 2012]. In this study, ALT levels more than three times the upper limit of normal were found in 1.1% of patients (2 of 176) receiving MLN + PGN compared with 0.6% (1 of 162) receiving PGN alone. Each treatment group also had one patient with an AST level more than three times the upper limit of normal. No other clinically relevant changes in laboratory values were found.
Discussion
This was the first study to examine the combined use of two approved medications in patients with fibromyalgia. The aim of this study was to evaluate the effects of adding MLN to PGN in patients with an incomplete response to PGN. Several factors support the rationale for this treatment approach. Although the efficacy of each drug has been demonstrated in large, placebo-controlled clinical studies [Arnold et al. 2008, 2010a; Crofford et al. 2008; Mease et al. 2008, 2009b; Clauw et al. 2009], not all patients with fibromyalgia are expected to fully respond to either PGN or MLN alone due to the complex nature of this disorder. Given the complementary mechanisms of action of these drugs, the addition of MLN (a serotonin and norepinephrine reuptake inhibitor) to PGN (an α2-δ ligand) may be beneficial in some of these patients who require more than one medication to treat the multiple symptoms of fibromyalgia [Mease and Seymour, 2008]. Moreover, as discussed in further detail below, an examination of the side-effect profile of these medications in combination may be useful.
In this study, incomplete responders were defined as patients meeting all of the following criteria: moderate to severe pain (VAS pain score ≥ 40 and ≤ 90); overall disease status reported as moderately to extremely ill (PGI-S score ≥ 4); and overall change with run-in PGN treatment rated as minimally improved, no change, or worsened (PGIC score ≥ 3). Of the 705 patients who received at least one dose of PGN during the run-in period, 101 (14.3%) were classified as responders and did not continue the study; 364 (51.6%) were classified as incomplete responders and continued into the randomized, open-label treatment period.
Results from the randomized population indicate that in patients experiencing an incomplete response to PGN, the addition of MLN improved patient global status, pain, and other symptoms associated with fibromyalgia. Based on primary and sensitivity analyses for the ITT population, approximately 50% of patients receiving MLN + PGN rated their global improvements from screening as ‘very much improved’ or ‘much improved’ compared with approximately 25% of patients receiving PGN alone (all analyses, p < 0.001). At first glance, the 25% responder rate in patients continuing PGN alone and not having any change in their medication appears to be rather high, particularly in view of the fact that they had very little change in their pain. One possible explanation is that these patients, who were incomplete responders after the run-in period, experienced some additional treatment benefits after 11 weeks of continued PGN treatment during the randomized, open-label period. In addition, and perhaps most importantly, it should be noted that to become a responder, patients only had to shift slightly over the boundary between ‘minimal improvement’ in PGIC (score of 3) during the run-in period with PGN and ‘much improvement’ (score of 2) to be deemed responders; thus, a relatively small degree of additional improvement could result in being classified as a responder.
At the end of the study, the percentage of patients with clinically meaningful pain improvements was approximately twice as high with MLN + PGN (45.8%) than with PGN alone (19.7%). Significant differences between treatment groups in mean pain scores were found at all randomized study visits (all visits, p < 0.001 versus PGN). In addition to these pain improvements, adding MLN to PGN resulted in significantly greater improvements in physical and mental functioning, fatigue, and cognition (all measures, p < 0.05 versus PGN).
Although rescue treatment with short-term opioids was allowed (except for within 7 days prior to scheduled study visits), the actual use of opioids in the study was low and unlikely to affect study outcomes. Vicodin was the most commonly used opioid in the randomized safety population (3.9%), reported more frequently in the PGN only group (5.6%) than in the MLN + PGN group (2.2%). The reasons for opioid prescription, time of initiation, and treatment duration were not analyzed.
Monitoring of safety and tolerability was an important aim of this study. No unexpected AEs were found with either PGN alone or MLN + PGN. The types of treatment-emergent AEs that occurred during the PGN run-in phase were consistent with previously reported results for this drug [Pfizer Inc., 2012]. As expected, these AEs did not generally worsen over time, as indicated by the lower incidences of treatment-emergent AEs for PGN alone during the randomized period compared with the PGN run-in period.
The addition of MLN to PGN was generally well tolerated, and did not appear to exacerbate any of the treatment-emergent AEs commonly associated with PGN or MLN monotherapy. In placebo-controlled studies with MLN 100 mg/day, for example, the most frequently reported AEs in patients with fibromyalgia were nausea (35%), headache (19%), constipation (16%), insomnia (12%), dizziness (11%), and hot flush (11%) [Forest Laboratories, Inc., 2012]. In the present study, the incidences of these events were notably lower in patients who received MLN + PGN (nausea, 12.5%; headache, 9.2%; constipation, 9.8%; insomnia, 5.4%; dizziness, 7.6%; and hot flush, 6.0%). In addition, rates of withdrawal due to nausea (2.7%) and palpitations (1.6%) in patients receiving MLN + PGN were lower than previously reported for MLN alone (nausea, 3.5%; palpitations, 2.6%) [Forest Research Institute, Inc., 2007]. Although the incidence of fatigue in patients receiving MLN + PGN (10.3%) was higher than found with MLN 100 mg/day in previous placebo-controlled trials (6.4% versus 6.0% for placebo), it was similar to the incidence reported with patients receiving PGN during the run-in period of this study (9.1%).
In patients receiving MLN + PGN, mean increases in blood pressure and heart rate from randomization were similar to vital sign increases found with MLN monotherapy [Forest Laboratories, Inc., 2012]. To evaluate the effects of run-in and randomized treatments on blood pressure and heart rate, changes from screening were also analyzed, and the results suggest the combined effects of PGN and MLN on blood pressure and heart rate may be somewhat less than the effects of MLN alone.
The main limitation of this exploratory study is the open-label design. Without a blinded placebo-control group, it is not possible to ascertain the extent to which improvements with combination therapy may have been due to patient or investigator expectations (i.e. placebo effect). At the time that this study was conducted, there were no available data on the combination of MLN + PGN on which to base sample size and power calculations. Therefore, this was designed as an exploratory study to evaluate whether a large-scale, placebo-controlled trial was feasible. The study results offer some clinical information regarding the potential benefits of adding MLN to PGN in patients with fibromyalgia, indicating the need for further research into combinations with different drug classes. Another potential limitation is the relatively short duration of the randomized treatment period, which was 11 weeks. Although the efficacy of MLN appears to remain constant for at least 3 years [Arnold et al. 2013], no firm conclusions can be drawn regarding long-term benefits of MLN + PGN. Finally, since the aim of the study was to evaluate the effects of adding MLN to PGN in patients with an incomplete response to PGN, conclusions can only be drawn about this particular study population. Again, further studies would need to be conducted to evaluate the benefits of combining these treatments in the general fibromyalgia population. Individual patient responses to monotherapies, as well as to combination therapies of medications with different mechanisms of action, highlight the ongoing need to study potential biomarkers of fibromyalgia to improve diagnosis and develop more targeted treatment strategies [Dadabhoy et al. 2008; Ablin et al. 2009].
Conclusion
The results from this open-label, randomized study suggest that adding MLN 100 mg/day to PGN therapy improved the global status, pain, and other symptoms of fibromyalgia in patients who experienced an incomplete response to initial PGN monotherapy. No unexpected AEs were found in this study, and the combination of MLN + PGN did not appear to exacerbate any of the treatment-emergent AEs that are described in the respective product labels. In fact, the incidence of some side effects in patients receiving MLN + PGN was lower than previously found with MLN monotherapy [Forest Laboratories, Inc., 2012]. For some patients requiring more than one fibromyalgia medication, the addition of MLN to PGN may be a favorable treatment option.
Footnotes
Acknowledgements
The authors wish to thank all of the patients and investigators who participated in this study. The authors also thank Mildred Bahn at Prescott Medical Communications Group (Chicago, IL, USA) for medical writing assistance supported by Forest Laboratories, Inc.
Funding
This study was supported by funding from Forest Laboratories, Inc.
Conflict of interest statement
Dr Mease has received research/grant funding, consultation fees, and speaker honoraria from Forest Laboratories, Inc., Cypress Bioscience, Inc., Jazz Pharmaceuticals, Eli Lilly and Company, Pfizer Inc., and UCB, Inc. Dr Farmer has received research/grant funding and consultation fees from Forest Laboratories, Inc. Dr Gendreau was formerly an officer at Cypress Bioscence, Inc., one of the companies involved in the development of milnacipran for the management of fibromyalgia. Drs Palmer, Trugman, and Wang are full-time employees of Forest Research Institute, Inc., a subsidiary of Forest Laboratories, Inc, and own stock in that company.