
Abstract
Background:
Postmenopausal breast cancer survivors treated with neo/adjuvant chemotherapy often experience persistent declines in physical function that fail to resolve after treatment. One mechanism thought to underlie this lasting impairment is cellular senescence, a fundamental process of aging that contributes to frailty and functional decline. Chemotherapy induces senescence, and preclinical studies show that targeting senescent cells with senolytics can reduce inflammation and improve physical function. These findings have generated a strong interest in translating senolytic therapies to humans; however, no study to date has evaluated the effects of senolytics on physical function in postmenopausal breast cancer survivors.
Objective:
To evaluate the effects of targeting senescence with the oral senolytic agent fisetin on physical function in chemotherapy-treated postmenopausal breast cancer survivors.
Design:
A multicenter, phase II, randomized, double-blind, placebo-controlled trial.
Methods and analysis:
Eighty-eight postmenopausal women with early-stage, high-risk breast cancer who completed neo/adjuvant chemotherapy within the past 12 months and have diminished physical function, defined by a 6-minute walk distance (6MWD) <400 m, will be randomized 1:1 to receive either placebo or fisetin (20 mg/kg/day) on days 1–3 of a 14-day cycle for four cycles. The primary endpoint is the change in the 6MWD from baseline to end of treatment.
Ethics:
The study has been approved by the institutional review boards at participating sites.
Discussion:
This is one of the first studies to test whether targeting senescence with an oral senolytic agent, fisetin, can mitigate physical function decline in postmenopausal breast cancer survivors treated with neo/adjuvant chemotherapy. Promising results would provide the preliminary evidence needed to support a larger, confirmatory trial evaluating fisetin’s efficacy in this population. If successful, this approach could fill an important unmet clinical need, as no pharmacological therapies currently exist to prevent or treat chemotherapy-related declines in physical function among postmenopausal breast cancer survivors.
Trial registration:
ClinicalTrials.gov NCT05595499.
Plain language summary
Many postmenopausal patients who complete chemotherapy for early breast cancer experience functional decline and don’t ever bounce back. Unfortunately, there are no approved pharmacological therapies that have been proven to prevent or reverse these long-term adverse effects of chemotherapy. We are therefore conducting this study to test an intervention to improve function in postmenopausal women who have functional decline after chemotherapy for early breast cancer. We are studying a supplement called fisetin, a naturally occurring substance found in strawberries and other fruits. Previous studies suggest that fisetin can get rid of cells in the body that have undergone a process called cellular senescence, where a cell becomes damaged but does not die. If these cells accumulate, they can lead to inflammation and loss of function. Studies have shown that chemotherapy can cause a rapid build-up of senescent cells. In mice, fisetin gets rid of senescent cells and improves physical function. In humans, fisetin is safe and reduces markers of senescence in various tissues. However, we do not know if fisetin can improve physical function in postmenopausal patients treated for early breast cancer. This research study will determine if fisetin can improve physical function in postmenopausal women with early-stage breast cancer with functional decline after treatment. Participants in this study will receive either fisetin or a placebo. To measure physical function, we will record how far participants can walk in six minutes before, during, and after the study. We will also perform other tests of function, collect health-related questionnaires, and obtain blood samples to look at blood markers of senescence. At the end of the study, we will be able to figure out if fisetin was more effective than placebo at improving the six minute walk distance. Promising results from this study will guide bigger studies to test fisetin in postmenopausal survivors of breast cancer.
Keywords
Introduction
Advances in breast cancer treatment have improved survival, 1 yet treatments used to cure the disease can accelerate age-related physical function decline.2–4 Indeed, approximately one-third of postmenopausal survivors treated with chemotherapy experience substantial, often persistent, loss in physical function.5–9 Postmenopausal survivors have a 2- to 4-fold increased risk of functional decline.5–9 While exercise can improve physical function after chemotherapy, the benefits are inconsistent, and many survivors struggle to recover with exercise alone.10–12 No approved pharmacological therapies currently exist to prevent or mitigate chemotherapy-related declines in physical function. Hence, there is an unmet need to develop safe, mechanism-based pharmacological interventions to restore and preserve physical function in cancer survivors.
One mechanism thought to drive chemotherapy-related physical function decline is cellular senescence. Senescence is a state of essentially irreversible growth arrest that arises in response to stress; it is a fundamental biological process of aging and is believed to be a key driver of age-related diseases, frailty, and functional decline.13–17 As we age, we accumulate senescent cells (Sncs) and many of these accumulated, persistent Sncs develop a senescence-associated secretory phenotype (SASP), releasing pro-inflammatory cytokines, chemokines, and other factors that promote tissue injury, fibrosis, and immune dysfunction.18–20
Senescence is triggered primarily as a response to damage, allowing suppression of potentially dysfunctional cells. Damage from cancer treatments, such as chemotherapy, has been shown to induce senescence. 21 Breast cancer survivors treated with adjuvant chemotherapy have increased peripheral blood T-cell expression of p16INK4a (p16), a known marker of Sncs, compared to those not receiving chemotherapy. 22 Higher p16 expression is linked with chemotherapy-related frailty, fatigue, and neuropathy21–24 and shown to persist for 12 months after treatment. 22 Seminal studies in animal models demonstrated that by expressing an inducible suicide gene under the control of the p16INK4a promoter, it is possible to ablate Sncs.25,26 The elimination of Sncs improved age-related diseases and dysfunction, delayed tumor formation, and ameliorated many side effects of chemotherapy.21,26,27 These findings have generated a strong interest in testing interventions that target and eliminate Sncs in cancer patients.
Small molecules called senolytics, which inhibit senescent cell anti-apoptotic pathways, have been proposed to selectively eliminate Sncs and, ultimately, reduce inflammation and promote tissue regeneration and healthspan.28,29, 44 First-generation senolytics are composed of repurposed products with known safety profiles, such as the natural flavonoid fisetin. Fisetin, a naturally occurring flavonoid found in fruits and vegetables, exhibits senolytic properties30–33 and enhances physical function in aged mice.32,34–36 In preclinical and early human studies, fisetin can be administered intermittently, with a “hit and run” dosing,16,28,32,37 and intermittent fisetin shows promise in reducing SASP markers; fisetin is also feasible, safe, and well-tolerated, with no reported serious adverse effects.38–42 However, whether fisetin can reduce Sncs and, in turn, improve physical function in postmenopausal breast cancer survivors has not been studied.
To address this knowledge gap, we designed the TROFFi study to evaluate the effect of the oral senolytic agent fisetin on physical function in postmenopausal breast cancer survivors treated with neo/adjuvant chemotherapy and experiencing diminished physical function.
Methods
Trial design
The TROFFi (Treatment Of Frailty with Fisetin) trial is a multicenter, phase II, randomized, double-blinded, placebo-controlled trial testing the effects of fisetin in chemotherapy-treated postmenopausal breast cancer survivors. TROFFi is an ongoing, federally-funded study that is currently open and recruiting participants at two large cancer centers, the University of California, Los Angeles (UCLA) and City of Hope (COH). The overall study design is shown in Figure 1.

Overview of the TROFFi study design. This study will randomize postmenopausal women with stage I–III breast cancer within 12 months of treatment with neo/adjuvant chemotherapy who have a 6-minute walk distance <400 m to either fisetin or placebo. The primary, secondary, and exploratory endpoints are listed.
Briefly, we will recruit 88 postmenopausal women with early-stage, high-risk breast cancer who have completed neoadjuvant or adjuvant chemotherapy and are experiencing diminished physical function, defined as having a 6-minute walk distance (6MWD) <400 meters (m), at the time of enrollment. Enrolled participants will be randomly assigned 1:1 to receive either a placebo or fisetin (20 mg/kg/day) on days 1–3 of a 14-day cycle for four cycles (Figure 2). The TROFFi protocol has been reviewed and approved by the institutional review boards at each participating site. Additionally, the reporting of this study conforms to the SPIRIT guidelines as detailed in the Supplemental Appendix. 43

Study schema outlining the timeline of events during the study period. After informed consent, participants will receive the study drug on days 1–3 of each 14-day cycle for four cycles. Study visits and research labs will be completed on the first day of cycle 1, cycle 3, and end of treatment. A safety follow-up will occur 30 days after end of treatment. Yearly chart reviews will be completed to follow up on recurrence and survival.
Eligibility criteria
Inclusion criteria
Postmenopausal women, diagnosed with stage I–III breast cancer who are within 30 days to 12 months of completing their last day of neo/adjuvant chemotherapy, are eligible for the study. Patients treated with chemotherapy who developed toxicity-related dose reductions, dose delays, or early discontinuation are included. Participants must also have evidence of diminished physical function (6MWD <400 m). Detailed inclusion criteria are presented in Table 1.
Inclusion criteria for TROFFi.
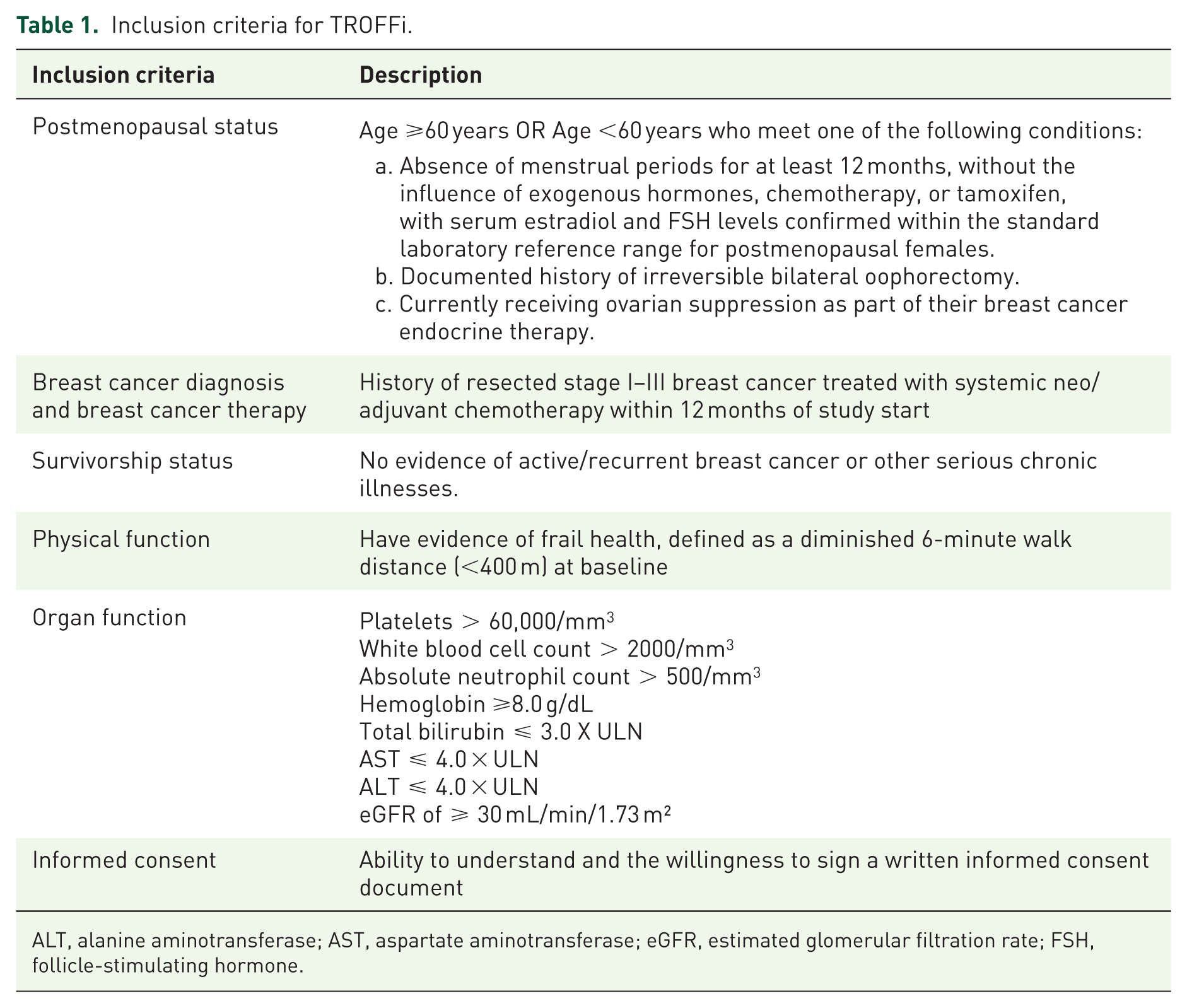
ALT, alanine aminotransferase; AST, aspartate aminotransferase; eGFR, estimated glomerular filtration rate; FSH, follicle-stimulating hormone.
Exclusion criteria
Exclusion criteria include recent cancer-directed therapy, major surgery, or radiation within 30 days prior to study treatment initiation; use of medications with potential or known senolytic effects; anticoagulants; cytochrome P-450 (CYP) drugs with a narrow therapeutic range; difficulty swallowing oral medications; active participation in another functional status study; and any condition deemed by the investigator to interfere with study participation. Participants who take concomitant medications that interact with CYP2C9, 2C19, and 1A2 will have their medications adjusted to be taken 10 h before or after fisetin administration on dosing days. These select medications are specified in the investigator’s brochure for fisetin. More detailed exclusion criteria are presented in Table 2.
Exclusion criteria for TROFFi.

Study recruitment and informed consent
Study recruitment will be conducted by research staff at UCLA and COH. Participants will be English or Spanish speaking. Strategies to identify potential participants include screening weekly lists of participants scheduled to be seen in outpatient oncology clinics, displaying study brochures in the waiting areas of participating clinics, obtaining physician referrals, and leveraging the electronic medical record to generate a registry of participants on chemotherapy regimens for early breast cancer.
Once potential participants are identified, study staff will conduct a pre-screening evaluation by asking participants to answer three questions. “Since your last chemotherapy treatment, (1) have you been experiencing fatigue or a decrease in energy levels? (2) have you been feeling weaker, slower, or less active than usual? and (3) have you lost more than 10 pounds unintentionally?” Participants who respond “yes” to any of these will pre-screen as potentially eligible and, if interested in the study, staff will then conduct a screening visit, during which the patient will be assessed for eligibility based on the 6MWD. If the participant’s 6MWD is <400 m, a study staff will review the study and, if the patient is interested, will obtain informed consent.
Randomization and blinding
After completing screening assessments, eligible participants will be randomized 1:1 to receive fisetin or placebo. Randomization will be stratified based on baseline 6MWD (<300 m vs >300 m), study site (UCLA vs COH), and hormone receptor (HR) status (HR positive vs HR negative) to ensure balanced distribution across treatment groups. The study investigators, research staff, and participants will be blinded to the randomization process and will remain blinded until the accrual goal is achieved and all participants complete the study.
Treatment program
Fisetin or placebo will be given for day (D) 1–3 of each cycle for a total of four cycles (C). Fisetin will be given at a dose of 20 mg/kg/day. This dose is adapted from effective senolytic activity in preclinical studies 38 and represents the current dosing of fisetin in multiple ongoing trials that are testing it as an oral senolytic agent in aging research. 45 Additionally, this dose and schedule have not been associated with any significant toxicity from these ongoing trials, consistent with prior published data on fisetin in humans.40,41
We obtained an IND from the US Food and Drug Administration to test fisetin that is produced from Sharp Clinical Services, a Good Manufacturing Practices facility. The fisetin supply for this study is funded by the National Institutes of Health, and participants will not bear any of the cost of the study agent. The fisetin/placebo will be provided in 100 mg capsules, and the total number of capsules will be determined based on the participant’s body weight. Participants will be weighed at the beginning of each study visit to determine the dosing weight. Weight will be rounded to the nearest kilogram, and the final dose will be rounded to the nearest 100 mg as this is the dose of each individual fisetin capsule. These small adjustments in dosing are not expected to change efficacy or have additional pharmacokinetic implications, as they will be <5% of the total administered dose. For example, a participant weighing 64 kg will require a 1280 mg/day dose of fisetin, which would translate to an administered dose of 1300 mg.
The first dose will be administered at the C1D1 visit and observed by the study staff. Participants will be encouraged to ingest all capsules of the study agent within 60 min, at approximately the same time each day to promote adherence. During each 2-week cycle, participants will take the study agent on D1 through 3 and will remain off the study agent from D4 through 14. Participants will complete a pill diary log to document adherence to the dosing regimen on D1-3 of each cycle. A window of up to 3 days will be allowed for scheduled visits to accommodate any unforeseen circumstances. Additionally, a 2-day window is given for pill intake if necessary to optimize adherence.
Study assessments
After screening, there will be four subsequent visits (baseline C1D1, C3D1, end of treatment [EOT], and safety follow-up), during which the following study assessments will be performed. The primary endpoint of the trial is the 6MWD, a well-established, accurate, sub-maximal exercise measure of aerobic capacity and endurance. Participants walk at their own pace for 6 min, and the distance (in meters) is measured at the end. The 6MWD will be measured prior to dosing of fisetin/placebo on C1D1 and C3D1 visits. We will also perform the 6MWD at the EOT and safety follow-up visits.
Beyond the 6MWD, at each study visit, we will collect other objective and subjective measures of physical function, including the Short Physical Performance Battery (SPPB)4,46,49 and the physical function component of the Short Form-36 (SF-36) (Table 3). 50 The SPPB includes a standing balance test, a gait-speed (4-meter walk) test, and a chair stand test. 49 The physical function component of the SF-36 is a patient-reported outcome that assesses limitations in physical activities, from basic tasks like bending or walking to more vigorous activities.51,52
TROFFi study questionnaires.

In addition to these efficacy endpoints of physical function, safety, tolerability, and adherence will also be assessed at each visit. Adverse events will be collected throughout the study using the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 50 as well as patient-reported outcome CTCAE (PRO-CTCAE). 51 Adverse events will be collected and documented at each study visit and at the safety follow-up. Any serious adverse events will be reported to the Data Safety Monitoring Board (DSMB) in writing within 10 days of awareness as specified by their protocol. Any deaths will be reported within 48 h. Adherence will be measured using a pill diary and collected from baseline to EOT.
Exploratory physical function endpoints will include grip strength 52 and Fried Frailty Phenotype. 53 Grip strength will be obtained using a hand dynamometer three times with each hand, and the best of the six grip strength measurements (in kilograms) will be used as the final measure. 52 Fried Frailty Phenotype is assessed using five criteria: unintentional weight loss, weakness, exhaustion, slow gait speed, and low physical activity; patients are then categorized as frail/prefrail or robust. Additional exploratory endpoints, including assessments of psychological function (anxiety, depression), cognitive function, quality of life, sleep, and chemotherapy-induced peripheral neuropathy, will be obtained using a series of self-reported electronic questionnaires completed by participants at their clinic visits (Table 3). Peripheral blood for biomarker studies will also be collected at baseline, C3D1, and EOT. Participants will not bear any costs of non-standard of care study assessments or research labs.
Study endpoints
The primary endpoint is the change in 6MWD from baseline to EOT. We selected the 6MWD as the primary endpoint for several reasons. First, the 6MWD has been demonstrated to measure treatment benefits in other diseases, and the FDA has accepted the 6MWD as a primary efficacy endpoint for pharmacological therapies in these diseases, including chronic obstructive pulmonary disease, pulmonary hypertension, and peripheral artery disease.54–58 Second, the 6MWD highly correlates with VO2peak, a measure of cardiopulmonary fitness and a marker of physiologic reserve that correlates with mortality. Patients with a 6MWD <400 m have an increased risk of all-cause mortality, and therefore by selecting for this group of survivors, the study aims to identify patients most likely to benefit from the proposed intervention. 59 Third, the 6MWD is expected to be sensitive to the proposed study interventions, as a previous study showed that senolytics may improve this outcome. 60
Secondary endpoints include changes in the SPPB and the physical function component of the SF-36 from baseline to EOT. Safety endpoints include clinician- and patient-reported adverse events, as measured by NCI CTCAE and PRO-CTCAE, and adherence obtained from pill diary for each cycle from baseline to EOT.
Exploratory endpoints include grip strength, frailty (Fried Frailty Phenotype), anxiety (GAD-7) and depression (PHQ-8), fatiguability (Borg RPE), cognition (PROMIS cognitive function short form), quality of life (SF-36), sleep (ISI), neuropathy (QLQ-CIPN20), as well as change in peripheral CD3+ T-cell p16 expression levels and circulating SASP inflammatory markers from baseline to EOT. Disease-free survival and overall survival endpoints will be assessed over 3 years following EOT.
Statistical methods
Sample size calculation
We used PASS 61 for sample size calculation, using an analysis of covariance framework, a test for a difference in means at follow-up controlling for the baseline measurement of the outcome variable. With 88 patients randomized 1:1 to fisetin or placebo, for a total of 44 patients per group, stratified by baseline 6MWD (<300 m vs >300), site (UCLA vs COH), and HR status (HR positive vs HR negative), and assuming a correlation of 0.4 between 6MWD at baseline and EOT, and 10% attrition by EOT, we will have 80% power to detect a standardized mean difference between treatments at EOT of 0.60, at significance level 0.05 two-sided. A standardized mean difference is the difference between groups in mean 6MWD divided by the common standard deviation (SD).
A meta-analysis of 1084 breast cancer survivors (mean age, 52 years) reported mean ± SD for 6MWD of 477.4 ± 75. 59 Given a SD of 75, a standardized mean difference of 0.60 corresponds to a treatment group difference in mean 6MWD of about 45 m. There are data to suggest that a minimal clinically important difference in the 6MWD in patients with breast cancer after completing chemotherapy to be approximately 40 m. 62 This effect size was also selected based on previously reported clinically important differences in 6MWD in non-breast cancer patient populations. Studies in other populations suggest a difference of 30 m to be clinically meaningful, while a change greater than 45 m is considered substantial. 63 With the selected sample size, we will have an 80% chance of concluding fisetin is efficacious if it improves the mean 6MWD by 45 m or more in the fisetin group compared to the placebo group.
Primary endpoint analysis
The primary endpoint is the difference in change over time, from baseline to EOT, in the 6MWD, compared by treatment arms. The analysis will be conducted as an intention-to-treat analysis, including participants in the arms to which they were randomized regardless of treatment received. 6MWD will be treated as a continuous variable, and higher 6MWD values indicate better physical function. Its distribution will be transformed to normality if necessary.
For the outcome analysis, we will use a linear mixed model with the repeated assessments of 6MWD as the dependent variable vector. The fixed effects will be assessment time point, study arm, and the interaction between time point and study arm, as well as the stratification factors used in the randomization. Time will be treated as a categorical variable, using indicator variables, to allow for non-linear trends. A random intercept for participant will be included to account for correlation of measurements within participant. The primary endpoint will be tested using a Wald test for the coefficient of the interaction between treatment and time (EOT). If it is significant at p < 0.05 two-sided and the mean change in 6MWD from baseline to EOT is higher for the fisetin arm than for the placebo arm, fisetin will be considered efficacious. We will compare baseline patient characteristics by arm, although imbalance is not anticipated due to randomization. If an imbalance is detected, we will conduct the analysis adjusting for covariates in the imbalance and conduct sensitivity analyses excluding the potential confounders.
Secondary efficacy and safety endpoint analysis
Secondary endpoints include the change in two other physical function measures beyond the 6MWD: the SPPB and the physical function component of the SF-36. Both endpoints are continuous measures and will be analyzed to assess differences in change over time between the fisetin and placebo groups in the same manner as described for the primary endpoint of 6MWD. To control the family-wise Type I error rate at 0.05 for these two secondary endpoints, we will use the Holm method. We will also perform cross-sectional correlations between the 6MWD and the SF-36 at each time point to evaluate the relationship between objective and subjective changes in physical function during the study.
Adverse events will be determined at each time point per patient as the presence (yes/no) of toxicities (CTCAE v5.0) of grade ⩾2. The number of patients with adverse events will be compared by treatment arms using Fisher’s exact test. Mixed models for binary data will also be used to compare the proportion of patients with adverse events over time by treatment.
Treatment adherence (yes/no) for each patient at each time point will be determined. A patient will be considered adherent if she took all the required capsules and non-adherent otherwise. We will then compare between treatments the proportion of adherent patients at each time point using linear regression models, adjusting for the stratification factors. Time point-specific analysis will be performed since conditions for pill-taking differ between the clinic (supervised) and patients’ home (self-administered).
Exploratory endpoints analysis
Exploratory endpoints include grip strength, frailty (Fried Frailty Phenotype), anxiety (GAD-7) and depression (PHQ-8), fatiguability (Borg RPE), cognition (PROMIS cognitive function short form), quality of life (SF-36), sleep (ISI), and neuropathy (QLQ-CIPN20). Local and distant breast cancer-free survival, breast cancer-specific survival, and overall survival will be compared between fisetin and placebo using the stratified log-rank test. The stratified Cox regression model will be used to obtain an estimate of the hazard ratio and the 95% confidence interval.
Analyses of p16INK4a and SASP markers will evaluate whether fisetin is associated with change in these markers (using mixed models as described for the primary endpoint); whether change in these markers is associated with change in physical function measures (by computing correlation coefficients for change scores); and whether higher baseline levels of these markers are associated with greater benefit (by testing interactions between baseline levels and treatment condition indicator). The distribution of continuous variables will be examined graphically, and appropriate transformations will be made before applying analytical methods based on the normal assumption.
Missing data
We expect attrition to be low (10% or less). We will collect and report dropout reasons, including elements such as breast cancer recurrence, development of a new cancer, need to start prohibited medication, worsening physical function, inability to attend study visits, and participant loss of interest. We will further compare participants who complete the study with those who do not on baseline characteristics to assess the potential for bias and perform multiple imputation analyses under missing at random and missing not at random assumptions if potential bias is suggested. The model will include all variables used in the primary and secondary analyses, including baseline characteristics such as age, race/ethnicity, cancer stage, study site, HR status, HER2 receptor status, and chemotherapy regimen.
Data management
Study data will be recorded within a Research Electronic Data Capture (REDCap) database. Only deidentified data with no patient health information will be exported for analysis. A fully de-identified (of all protected health information) database will be generated and used for statistical analysis as well as monitoring by the DSMB.
Discussion
Physical function decline after chemotherapy remains a pervasive and debilitating problem for postmenopausal breast cancer survivors, yet no approved, mechanism-based pharmacologic therapies currently exist to prevent or reverse it. To our knowledge, this study is the first to test whether fisetin—a natural flavonoid with senolytic properties—can target cellular senescence, a biological process thought to drive the accelerated aging effects of chemotherapy and improve physical function in postmenopausal breast cancer survivors. Positive results from this phase II study could generate the foundational evidence needed to support a larger, multicenter confirmatory trial and help establish the efficacy of fisetin for chemotherapy-related functional decline in this population. If effective, fisetin could address a critical unmet need, offering one of the first pharmacologic options to prevent or treat chemotherapy-related declines in physical function among postmenopausal breast cancer survivors.
This research is important and innovative for three key reasons. First, this phase II study introduces a mechanism-based strategy to address chemotherapy-related functional decline by targeting cellular senescence—a biological hallmark of aging not previously studied in this clinical context. Targeting senescence, an upstream driver of frailty and functional decline, holds considerable therapeutic promise and establishes a prototype for future studies aimed at mitigating cancer treatment–related aging processes, including physical, cognitive, and psychological impairments. Second, fisetin is a novel, low-toxicity, orally administered intervention that is widely available and well tolerated, making it a practical candidate for integration into survivorship care. The study design is pragmatic and clinically oriented, facilitating straightforward adoption and implementation if successful. Third, by focusing on physical function as a measurable and meaningful endpoint, this study prioritizes patient-centered outcomes that directly influence independence, quality of life, and overall health. Together, these features position this work to advance the development of precision, biology-driven interventions that could be paradigm-shifting in how we approach cancer survivorship.
Our study will also examine the effect of fisetin on subjective measures of physical function and evaluate the relationship between the 6MWD and subjective measures such as the SF-36. If we find that the 6MWD improves but the subjective measures do not, we will hypothesize that the improvement was not immediately clinically significant from a quality of life and symptom perspective. If we find that the 6MWD does not improve, but the subjective measures do, we will hypothesize that the 6MWD may not be sensitive enough to detect meaningful change in this setting and use this to guide future studies. Furthermore, we are including other measures of health beyond physical function such as cognitive and psychological function, sleep, and quality of life. Similar to physical function, these facets of health are often negatively affected by chemotherapy, and there are limited evidence-based interventions that address these issues.67–71 Positive data on these endpoints will guide future studies looking at the impact of senolytics on chemotherapy-related complications in both survivors of breast cancer and other cancer types where chemotherapy remains a backbone of treatment.
A key goal of this study is to determine whether fisetin meaningfully reduces Snc burden, as reflected by changes in circulating p16INK4a expression and SASP factors. Although these analyses are exploratory, they will allow us to link biological effects with clinical outcomes by exploring whether reductions in senescence markers correlate with improvements in physical function. We anticipate that fisetin will decrease markers of senescence and inflammation, supporting its hypothesized mechanism of action. However, responses may vary; certain participants with higher baseline senescence burden may derive greater benefit, while others may show minimal biomarker or functional change. Identifying these subgroups could inform future precision approaches and help tailor senolytic therapies to those most likely to respond.
This study also has several limitations. First, although we include both objective (e.g., 6MWD) and subjective (e.g., physical function component of SF-36) measures of physical function, we do not employ real-time monitoring tools such as wearable accelerometers that could capture daily activity and subtle, home-based changes in function. Future studies should incorporate digital health technologies to complement in-clinic assessments and provide a more continuous measure of functional recovery. Second, we rely on the 6MWD as a surrogate of physiological reserve rather than a more integrative cardiorespiratory fitness test to measure VO2peak; subsequent studies could include cardiopulmonary exercise testing to validate findings and strengthen physiological interpretation. Third, the minimal clinically important difference for change in 6MWD has not been well established specifically for postmenopausal breast cancer survivors who have physical function decline after treatment. The current sample size is powered based on a minimal clinically important difference in 6MWD that is extrapolated from changes seen among patients with breast cancer before and after treatment as well as in other clinical populations, which may limit the interpretation of the clinical relevance of a positive finding. To address this, we will generate preliminary data on 6MWD responsiveness in this population, which can guide the design and powering of future confirmatory trials. Fourth, the 3-year follow-up period consists primarily of chart review to document disease-related outcomes and does not include longitudinal reassessment of physical function, which is beyond the scope of the current protocol and represents an area for future research. Building on this framework, future work could integrate remote or digital follow-up assessments to track long-term functional trajectories. Finally, it should be noted that our eligibility is limited to postmenopausal women. This restriction aligns with FDA guidance, as safety data on high-dose, intermittent fisetin—or other senolytic agents—are lacking in women of childbearing potential. Men were also excluded because sex may influence biological aging,69–73 and the small number of men with breast cancer would preclude meaningful analysis. However, future studies should expand to include both premenopausal women and men once adequate safety and dosing data are available, enabling examination of potential sex- and hormone-related differences in response to senolytic therapy.
Despite these limitations, this study has notable strengths. By utilizing a multicenter, randomized, double-blinded, and placebo-controlled design, we maximize internal validity and control for placebo effects, which is critical given that functional outcomes can be influenced by subjective factors. We have also prioritized minimizing participant burden without compromising data collection by reducing the number of in-person visits, enabling remote completion of questionnaires, and coordinating study visits with other clinical appointments whenever feasible. Furthermore, we selected fisetin, a natural flavonoid with senolytic properties, which has been shown to be safe and well-tolerated in humans.32,35,40 Fisetin’s favorable toxicity profile makes it a particularly suitable candidate for breast cancer survivors, a population at increased risk for treatment-related morbidity. By prioritizing safety alongside efficacy, we aim to maximize the therapeutic potential of senolytic therapy while minimizing additional burdens on this vulnerable patient population.
Ultimately, findings from the TROFFi trial could meaningfully inform our clinical approach for postmenopausal breast cancer survivors. If positive, this study would provide strong preliminary data and a rationale for larger confirmatory trials to establish fisetin as a novel, mechanism-based therapy for chemotherapy-related functional decline. Beyond its immediate clinical implications, TROFFi could help serve as a prototype for a new approach to cancer survivorship research—one that targets the biology of aging to help survivors not only live longer, but live stronger, maintain independence, and enjoy a better quality of life.
Supplemental Material
sj-docx-1-tam-10.1177_17588359261424668 – Supplemental material for A phase II randomized placebo-controlled study of fisetin to improve physical function in breast cancer survivors: the TROFFi study rationale and trial design
Supplemental material, sj-docx-1-tam-10.1177_17588359261424668 for A phase II randomized placebo-controlled study of fisetin to improve physical function in breast cancer survivors: the TROFFi study rationale and trial design by Jingran Ji, Catherine M. Crespi, Lisa Yee, Yuliya A. Zekster, Ali Al-Saleem, Laura Petersen, Catherine Lee, Nala Son, Claire Smith, Tamara Evans, Tamar Tchkonia, James L. Kirkland, George A. Kuchel, Harvey Jay Cohen and Mina S. Sedrak in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We gratefully acknowledge the patients who generously contribute their time and experiences to make this research possible.
Authors’ note
The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.