
Abstract
Background:
Drug repurposing has emerged as an effective strategy to accelerate drug discovery. Using the pipeline established from a large collaborative drug repurposing project focused on high-grade serous ovarian cancer (HGSOC), we identified ivacaftor, an FDA-approved cystic fibrosis medication, as a drug candidate predicted to interact with the receptor tyrosine kinase-like orphan receptor 1 (ROR1) which we have previously demonstrated as a therapeutic target in ovarian cancer.
Objectives:
This study aimed to provide preclinical evidence supporting the potential repurposing of ivacaftor for HGSOC treatment.
Design:
Ivacaftor was tested in 2D and 3D preclinical models as well as patient-derived organoid models in vitro.
Methods:
Dose–response analysis was undertaken in ROR1 expressing HGSOC cell lines OVCAR4, KURAMOCHI, COV362 and COV318 in both 2D adherent and 3D bioprinted formats. Real-time live/dead and apoptosis cell staining were performed over a 72 h period using the IncuCyte live cell imaging platform. Flow cytometry was used to assess apoptosis, DNA damage and cell proliferation following treatment with either 15 µM ivacaftor or 30 µM carboplatin at 24, 48 and 72 h. Additionally, ROR1-expressing HGSOC patient-derived organoids (OC029, OC043 and OC058) underwent ivacaftor dose–response analysis. Cell apoptosis following 15 µM ivacaftor treatment was measured in real-time using an Annexin V assay in two additional organoid models (OC062 and OC075). Finally, the mechanisms associated with response to ivacaftor were explored in HGSOC cell lines through Western blotting.
Results:
The IC50 for ivacaftor ranged from 6.5 to 13.2 µM in 2D cultures and 11.6 to 18.2 µM in 3D cultures. Treatment with 10 and 15 µM ivacaftor resulted in significantly increased cell death and reduced live cell counts compared to the vehicle control over 72 h. Organoids displayed IC50 values between 11.2 and 14.1 µM. Ivacaftor treatment induced apoptosis in organoids, with no significant impact on DNA damage or cell cycle in HGSOC cells. ROR1 signalling associated oncogenic pathways including the BMI-1 and the PI3K/AKT pathways were modulated following ivacaftor treatment.
Conclusion:
In summary, ivacaftor demonstrated significant anti-tumour potential in preclinical HGSOC models, supporting its further investigation as a repurposed therapy for ovarian cancer.
Keywords
Introduction
Ovarian cancer has been ranked the 8th most common cancer in women globally. 1 As the most common subtype, high-grade serous ovarian cancer (HGSOC) is associated with a high recurrence rate (~75%) and chemoresistance. 2 Characterised by TP53 mutations, HGSOC exhibits significant heterogeneity. 3 Standard treatment involves cytoreductive surgery with or without platinum and taxane-based chemotherapies, followed by maintenance therapy with a PARP inhibitor (PARPi) in patients with homologous recombination deficient (HRD) pathology. While PARPi and other targeted therapies have improved outcomes, not all patients respond. For patients characterised as HR proficient (HRP), who make up almost half of all HGSOC cases, the benefits of maintenance treatment with antiangiogenic agents or PARPi are limited, 4 highlighting the urgent need for novel therapies.
Drug repurposing is the process of identifying new therapeutic uses for existing or investigational drugs. This approach has gained significant attention in recent years as it offers several advantages over traditional de novo drug discovery, including reduced cost, shorter development timelines and lower risk of drug attrition. One promising target for drug repurposing is the receptor tyrosine kinase-like orphan receptor 1 (ROR1). ROR1 has been implicated in the pathogenesis of various types of cancer, including HGSOC.5–8
ROR1 is a transmembrane protein that transduces signals within the non-canonical Wnt signalling cascade. 9 It plays a critical role in early embryogenesis but diminishes post-partum and is typically absent in healthy adult tissues. 10 However, ROR1 has been found to be aberrantly reactivated in several types of cancer and contributes to tumour cell growth, metastatic potential and chemoresistance.11–15 The oncogenic role of ROR1 has been associated with several downstream pathways including the Rho GTPases, epithelial to mesenchymal transition, PI3K/AKT, Hippo-YAP/TAZ and BMI-1.13,16–19 Promising preclinical evidence has led to the development and investigation of a number of ROR1 targeted therapies, several of which have advanced into early-phase clinical trials. These therapies primarily target the extracellular domain of ROR1, including the humanised monoclonal antibody zilovertamab (previously known as cirmtuzumab or UC-961),20,21 the antibody drug conjugate (ADC) NBE-002 22 (NCT04441099) and ROR1-specific chimeric antigen receptor T-cell therapy. 23 In contrast, an alternative ROR1-targeting strategy involving small molecule inhibitors that target the intracellular kinase domain of ROR1, such as KAN0441571C 24 and ARI-1 25 or other compounds with ROR1 inhibition activity such as strictinin 26 and GZD824 (also known as olverembatinib), 17 have demonstrated preclinical efficacy in ROR1 related haematological cancers and malignancies, including ovarian cancer. This study reports a comprehensive drug repurposing approach to explore the potential of targeting ROR1 using existing therapies for the treatment of HGSOC.
Methods
The reporting of this study conforms to the ARRIVE guidelines 2.0 27 (Supplemental Table 1).
In silico drug target analysis
Compounds with potential for broad-spectrum ROR1 binding activity were identified using the in silico BLAZE™ application developed by Cresset Discovery Services UK. 28 BLAZE is a ligand-based virtual screening platform which uses the shape and electrostatic character of ligands to rapidly search large chemical collections for molecules with similar properties. This method generated a preliminary list of compounds that included approved and investigational drugs. An in silico search was conducted to identify compounds which had FDA/EMA/TGA approval for use in any indication. Drugs identified through in silico BLAZE screening were filtered to exclude active pharmaceutical components that have not been FDA- or TGA-approved. The PRISM (Profiling Relative Inhibition Simultaneously in Mixtures) Repurposing Database 29 (Broad Institute, Cambridge, MA, USA) was used to further narrow down the most effective drugs across seven HGSOC cell lines. Both the 2019 and 2024 datasets were included for the screening. A search strategy was then implemented to rank the drugs based on properties such as lipophilicity (LogP) score (indicating lipid solubility), drug half-life, safety and toxicity profiles from other uses, achievable maximum plasma concentration (Cmax), clinical trial data for other cancers and in vitro evidence of cytotoxicity within the approved dosing window. Additionally, we incorporated feedback from our ovarian cancer consumer group to assess adverse events and to look for potential drug–drug interactions. The top 4 drugs from this pipeline, specifically ivacaftor, natamycin, cefoperazone and pirenzepine were selected for further investigation.
Drug preparation
Stock solutions of ivacaftor (100 mM, cat. #HY-13017), the antifungal natamycin (25 mM, cat. #HY-B0133) and the cephalosporin antibiotic cefoperazone (150 mM, cat. #HY-B0210) were prepared in dimethyl sulfoxide (DMSO), while the selective muscarinic antagonist pirenzepine (150 mM, cat. #HY-17037) was dissolved in sterile water. All compounds were sourced from MedChemExpress (Monmouth Junction, NJ, USA). The solutions underwent brief ultrasonication and were stored at −80°C.
In vitro screening – 2D HGSOC cell lines
Four HGSOC cell lines were used in this study; the KURAMOCHI (RRID: CVCL_1345), OVSAHO (RRID: CVCL_3114), OVKATE (RRID: CVCL_3110), TYKNU (RRID: CVCL_1776), COV362 (RRID: CVCL_2420), COV318 (RRID: CVCL_2419) and OAW28 (RRID: CVCL_1614) cell lines were supplied by the JCRB Cell Bank (National Institutes of Biomedical Innovation, Health and Nutrition, Japan), as catalogue numbers JCRB0098, JCRB1046, JCRB0234.0, 7071910, 7071903 and 85101601, respectively. The cell lines were purchased from CellBank Australia (Westmead, NSW, Australia). The cell lines have been previously described.30–33 These cell lines were confirmed by genomic and proteomic profiling to be representative of HGSOC34,35 and all four cell lines were shown to express the ROR1 transcript. 36 The cell lines were cultured in high glucose DMEM (Gibco; Thermofisher Scientific, Scoresby, VIC, Australia) plus 10% fetal bovine serum (Sigma-Aldrich; Merck, Darmstadt, HE, Germany) at 37°C in humidified 5% CO2. Cell line authentication was undertaken by the Australian Genome Research Facility, Melbourne, Australia. Mycoplasma testing was conducted using the MycoAlert™ Mycoplasma Detection Kit (cat. #LT07-318; Lonza, Walkersville, MD, USA). All cell lines were distributed by CellBank Australia.
Drug treatment and cell viability assay
HGSOC cell lines were plated and incubated for 24 h before being treated with increasing concentrations of the drugs for 72 h. Each drug was serially diluted (2-fold) in cell culture media to achieve the desired concentrations: ivacaftor and natamycin (0.39–100 µM), cefoperazone (3.91–1000 µM) and pirenzepine (0.98–250 µM). Cell viability after drug treatment was assessed using 0.4 mg/mL MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) that was added to each well and incubated at 37°C for 3 h. The media and MTT solutions were removed from the wells after 3 h and discarded. A total of 150 µL DMSO was added to each well and absorbance was measured at 570 nM in a SPECTROstar Nano plate reader (BMG Labtech, Ortenberg, BW, Germany). Three independent experiments were performed for each cell line. A dose–response curve was generated in GraphPad Prism to determine the IC50 value.
Live-cell analysis
HGOSC cell lines were plated and incubated at 37°C for 24 h, then treated with ivacaftor and carboplatin and incubated at 37°C for 5 days. IncuCyte® Cytotox Green (final concentration of 2.5 nM; Sartorius, Göttingen, LS, Germany) and Annexin V Red (final concentration of 1:400 of the reagent stock concentration; Sartorius, Göttingen, LS, Germany) were added to the cells during the drug treatment. IncuCyte Cytotox Green was used to stain dead cells and IncuCyte Annexin V Red was used to stain apoptotic cells. Cells were imaged in real time using IncuCyte SX5. Three independent experiments were performed for each cell line.
In vitro screening – 3D bioprinted HGSOC cell lines
The RASTRUM™ 3D bioprinting platform (Inventia Life Science, Sydney, NSW, Australia)37,38 was used to generate 3D cell models of COV318, COV362, KURAMOCHI and OVCAR4 utilising commercially purchased polyethene glycol-based ‘bioinks’ and crosslinking activators that polymerise to form hydrogel matrices (‘Px03.85’). Hydrogels were of an elastic modulus of 3.0 kPa and biofunctionalised with adhesion peptide motifs RGD (Arg–Gly–Asp), GFOGER (Gly–Phe–Hyp–Gly–Glu–Arg) and DYIGSR (Tyr–Ile–Gly–Ser–Arg) and full-length protein fibronectin. Cells were encapsulated into biofunctionalised hydrogels as a ‘large plug’ model and deposited onto bases of inert hydrogel in 96-well, tissue culture-treated, low evaporation plates (Corning, NY, USA). Printing densities utilised for cell lines were 5.56 × 106 cells/mL (16,000 cells/well) for COV318 and OVCAR4, 6.25 × 106 cells/mL (18,000 cells/well) for KURAMOCHI and 12.5 × 106 cells/mL (36,000 cells/well) for COV362. Cell densities were optimised depending on cell size and growth rate, allowing for 3D structures to be formed at 4 days from printing, permitting the addition of ivacaftor or DMSO vehicle control at this time point.
Bioprinted cell models were treated for 72 h with 10 concentrations of ivacaftor generated through 1.5-fold serial dilutions starting from 100 µM, or DMSO vehicle control. CellTiter-Glo®-3D bioluminescent reagent (Promega, Madison, WI, USA) was used as per the manufacturer’s instructions to assess cell viability following ivacaftor treatment, where luminescence was measured using a Tecan Infinite® M Plex plate reader (Tecan, Männedorf, ZH, Switzerland). Cell viability, computed as the average of duplicate values, minus background readings (wells containing inert base and media only), was expressed as a percentage relative to DMSO vehicle control.
In vitro screening – 3D HGSOC patient-derived organoids
Ovarian cancer tissue and ascites were collected with informed consent from patients (clinicopathological parameters shown in Supplemental Table 2). Upon receipt, the tumour specimens were dissociated into single cells via mechanical trituration and enzyme digestion with 2 mg/mL collagenase IV (Thermofisher Scientific). For the ascites fluids, tumour cells (primarily derived from the spheroids) were isolated using a 40 µm cell strainer. The isolated cells were embedded in 70% Matrigel/30% defined medium matrix and cultured in the defined medium (recipe provided in Supplemental Table 2 from Matthews et al. 39 ). For dose–response assays, organoids were harvested from Matrigel domes, dissociated using TrypLE (Thermofisher Scientific) and seeded into a 96-well plate in triplicates at a density of 8000 cells per well. After a 72 h establishment period, ivacaftor or vehicle control (0.1% DMSO) was added to each well in serial dilutions (0.78–100 µM), followed by a 72 h incubation. Cell viability was assessed at the endpoint using the CellTiter-Glo assay (Promega).
Flow cytometry analysis of apoptosis, DNA damage and cell cycle
The effect of ivacaftor on apoptosis, DNA damage and cell cycle was further determined by flow cytometry using the BD Pharmingen™ Apoptosis, DNA Damage and Cell Proliferation Kit (BD Biosciences, San Jose, CA, USA), as per the manufacturer’s protocol. Briefly, 3.5 × 105 cells/well of COV362 and KURAMOCHI cell lines were seeded into 6-well plates and incubated for 24 h. Cells were then treated with either DMSO vehicle control, 15 µM ivacaftor or 30 µM carboplatin as a positive control for 24, 48 or 72 h, and trypsinised after these points. Cells were fixed and permeabilised for 15 min at room temperature with BD Cytofix/Cytoperm™ Fixation/Permeabilisation Solution, a proprietary fixation and permeabilisation reagent containing a mixture of paraformaldehyde and saponin. The fixed cells were labelled with Alexa Fluor™ 647 Mouse Anti-H2AX (pS139) and phycoerythrin (PE) Anti-Cleaved PARP (Asp214) and incubated at room temperature for 20 min. Cells were then resuspended in 1 µg/mL DAPI in labelling buffer. The resuspended cells were analysed using a Fortessa X-20 flow cytometer (BD Biosciences). Data was analysed using FlowJo v.10 (FlowJo, Ashland, OR, USA). The median fluorescence intensity for Alexa Fluor 647 and PE was calculated for each sample and expressed as a ratio compared to the median fluorescent intensity of the DMSO control. The cell cycle distribution was analysed, and each cell cycle phase was displayed as a fraction of the total cells counted.
Quantitative reverse transcription polymerase chain reaction
ROR1 mRNA expression level in each HGSOC cell lines were analysed using real-time reverse transcription polymerase chain reaction (RT-PCR) as previously described. 40 Briefly, total RNA was extracted from the cells and reverse transcribed into complementary DNA using the QuantiTect Reverse Transcription Kit (Qiagen, Melbourne, VIC, Australia). ROR1 and three reference genes (HSPCB, SDHA and RPL13a) were amplified via PCR using QuantiNova SYBR green PCR kit (Qiagen) and primers (ROR1 forward: CAACAAGAAGCCTCCCTAATGG, ROR1 reverse: CCTGAGTGACGGCACCTAGAA; SDHA forward: TGGGAACAAGAGGGCATCTG, SDHA reverse: CCACCACTGCATCAAATTCATG; HSPCB forward: TCTGGGTATCGGAAAGCAAGCC, HSPCB reverse: GTGCACTTCCTCAGGCATCTTG; RPL13a forward: CCTGGAGGAGAAGAGGAAAGAGA, RPL13a reverse: TTGAGGACCTCTGTGTATTTGTCAA), on an AriaMX Real-time PCR System (Agilent, Santa Clara, CA, USA). The relative expression level of ROR1 was estimated using the 2−ΔΔCt method and normalised against the mean of the reference genes.
CRISPR/Cas9 mediated establishment of ROR1 knockout cell lines
ROR1 knockout (KO) cell lines were transfected with human ROR1 ef1a-puro all-in-one sgRNA lentiviral particles (Horizon Discovery, Lafayette, CO, USA; Cat# VSGH12608-256927277; target sequence: GTGCGTGGCAACAAACGGCA). Puromycin (Thermofisher Scientific) selection was performed using the concentration determined via antibiotic kill curve assays. Selection was maintained for 2 weeks to enrich for puromycin-resistant cells. Successful ROR1 knockout was confirmed by Western Blot analysis.
Western blotting
Protein expression of markers of interest were analysed using Western blotting as previously described. 17 In brief, total protein lysates were prepared using cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) with protease and phosphatase inhibitor cocktail (Cell Signaling Technology) and loaded in 4%–20% Mini-PROTEAN® TGX™ Precast Protein Gels (BioRad, Sydney, NSW, Australia). The blotted nitrocellulose gels were blocked in 5% non-fat milk (Coles, Australia) and incubated with primary antibodies overnight at 4°C, before being incubated with secondary antibodies for 1 h at room temperature. The chemiluminescent signals were developed using the SuperSignal West Atto kit (Thermofisher Scientific) and observed using the iBright Imaging system (Thermofisher Scientific). The primary antibodies used were ROR1 (R&D Systems, Minneapolis, MN, USA; #AF2000), ROR2 (QED Bioscience, San Diego, CA, USA; #34045), BMI-1 (Cell Signalling Technology; #6964), cleaved Caspase 3 (Cell Signalling Technology; #9661), cleaved PARP (Cell Signalling Technology; #5625), pAKT (Ser473; Cell signalling Technology; #4060), AKT (Cell Signalling Technology; #4691) and GAPDH (Cell Signalling Technology; #2118).
ROR1 immunohistochemistry
Immunohistochemistry (IHC) for ROR1 was performed at the Katharina Gaus Light Microscopy Facility at the UNSW Mark Wainwright Analytical Centre. Formalin-fixed paraffin-embedded slides were deparaffinised within the staining instrument and immunostained on a Bond Polymer Refine Detection System (Leica Biosystems, Melbourne, VIC, Australia). The mouse anti-ROR1 antibody (clone 6D4) 10 was diluted 1:9000. The signal was developed using the Bond Polymer Refine Detection system (Leica Biosystems) and counterstained with Haematoxylin.
Statistical analysis
Half-maximal inhibitory concentration (IC50) was estimated from dose–response curves generated with GraphPad Prism Version 10.4.0 software (GraphPad Software, San Diego, CA, USA) non-linear regression function. Fluorescence intensity of Alexa Fluor 647 and PE from the flow cytometry analysis was expressed as the mean ± SEM of each fluorescent label from the three replicate experiments for each cell line, unless otherwise specified. Results were compared to the DMSO vehicle and differences in these ratios at 24, 48 and 72 h were determined a one sample t test. Significance was set at p < 0.05. The proportion of cells in cell cycle phases were compared using a one-way ANOVA with Tukey’s post hoc test for multiple comparisons. Correlation between ROR1 mRNA level and IC50 values was analysed using the Pearson correlation test. Two-way ANOVA (time and treatment as two independent factors) was performed to compare different treatments and vehicle control, followed by post hoc testing with the Dunnett’s multiple comparisons test. Figures were generated using GraphPad Prism (v10.2.0). Data were displayed as mean ± SEM unless otherwise specified. Significance was set at p < 0.05.
Results
Approved drugs predicted to bind to ROR1 and have cytotoxic effects
In silico screening of all approved drugs from the British Pharmacopoeia with potential ROR1 binding activity was performed using the BLAZE software (Cresset Discovery Services, Litlington, Cambridgeshire, UK). Figure S1 showed the location of BLAZE screen ligand template between the ROR1-Kringle domain and the antibody heavy chain variable domain. A total of 253 compounds with potential ROR1 binding were identified. Of these compounds, 129 were approved for other indications, and 123 were under assessment in pre-clinical or clinical trial studies.
Of the 129 drugs already approved for other indications, there was data available for 71 drugs in 7 HGSOC cell lines in the Broad Institute PRISM Repurposing Database (Figure S2), 4 of which were used in this study. This list of 71 drugs was further refined to 8 drugs after considering drug properties including LogP, half-life, safety profile, Cmax and data from clinical trial and in vitro assessments. The eight drugs that met the criteria were natamycin, cefoperazone, ivacaftor, linagliptin, alogliptin, tipranavir, pirenzepine and trametinib (Figure 1(a) and (b)).

Top candidates shortlisted for ROR1-targeted therapies in HGSOC cell lines. (a, b) Log (2) fold-change of cell number after treatment with 2.5 µM dose of each of the top 8 drugs compared to DMSO in HGSOC cell lines from the Broad Institute PRISM repurposing datasets (2019 and 2024). (c–f) In vitro dose–response curves measured as a percentage of cell viability compared to untreated for natamycin, pirenzepine, cefoperazone and ivacaftor in HGSOC cell lines COV362 and OVCAR4. The dose–response curves displayed is mean ± SEM of triplicates of three independent experiments from combined analysis of COV362 and OVCAR4.
Four lead drugs with cytotoxic potential and low toxicity, specifically natamycin, cefoperazone, pirenzepine and ivacaftor, were first tested in vitro. HGSOC cell lines (COV362 and OVCAR4) were treated with increasing doses of each of the drugs to determine the IC50 value. The highest doses of drugs used to generate dose–response curves were 100 µM for natamycin, 1000 µM for cefoperazone and 250 µM for pirenzepine. As these doses were higher than the reported Cmax of the drugs (natamycin Cmax not available, cefoperazone Cmax = 581 µM, 41 pirenzepine Cmax = 41.6 µM) 42 (Figure 1(c)–(e)), they were determined to have low translational potential in HGSOC and excluded from further screening. Ivacaftor, an allosteric receptor modulator,43,44 was investigated in vitro as the lead candidate drug with potential to bind to ROR1 and exhibit cytotoxic effects (Figure 1(f)).
Ivacaftor has cytotoxic effects in in vitro ROR1-positive models of HGSOC
Four ROR1-positive HGSOC cell lines were treated with increasing concentrations of ivacaftor and the cell viability determined using MTT colorimetric assay. Relative ROR1 mRNA levels normalised to reference genes for each of the cell lines were COV318 (1.56 ± 0.28), COV362 (0.41 ± 0.04), KURAMOCHI (0.44 ± 0.05) and OVCAR4 (0.09 ± 0.01). Ivacaftor dose–response curves for each cell line were generated and the IC50 values (dose at which cell viability was reduced to 50%) determined. Ivacaftor was shown to have a cytotoxic effect across all four cell lines (Figure 2, IC50 = 10.56 ± 2.89 µM). A negative correlation was observed between ROR1 mRNA levels and the IC50; however, this was not statistically significant (Figure 2(e), r = −0.90, p = 0.10).

HGSOC cell lines in 2D are sensitive to the cytotoxic effects of ivacaftor. Dose–response curves and IC50 values for ivacaftor in four HGSOC cell lines (a) KURAMOCHI, (b) COV362, (c) OVCAR4 and (d) COV318. The data displayed is mean ± SEM of triplicates of three independent experiments. (e) Correlation between ROR1 mRNA level and IC50 values for ivacaftor of the HGSOC cell lines. Relative ROR1 mRNA levels for each of the cell lines normalised to reference genes were COV318 (1.56 ± 0.28), COV362 (0.41 ± 0.04), KURAMOCHI (0.44 ± 0.05) and OVCAR4 (0.09 ± 0.01).
The cytotoxic effects of ivacaftor over time were assessed by measuring HGSOC cell growth after treatment with doses of ivacaftor similar to the mean (10.56 µM) and maximum (13.24 µM) IC50 values of all cell lines. HGSOC cell lines were treated with 10 and 15 µM of ivacaftor, and 30 µM (plasma Cmax 45 ) and 100 µM of carboplatin for comparison to a standard treatment for HGSOC (clinically relevant concentration 46 ). The cell growth, death and apoptosis in HGSOC cells treated with ivacaftor or carboplatin compared to vehicle control was assessed by live-cell analysis (Figure 3). For three out of four cell lines, 15 µM of ivacaftor significantly reduced cell growth, increased cell death and apoptosis compared to vehicle control. Specifically, in COV318, COV362 and OVCAR4 cell lines, 15 µM ivacaftor induced greater apoptosis than carboplatin.

Real-time cell monitoring following ivacaftor and carboplatin treatments. Cell growth (a, d, g, j), death (b, e, h, k) and apoptosis (c, f, i, l) of KURAMOCHI, COV362, COV318 and OVCAR4 cell lines treated with ivacaftor (10 or 15 µM), carboplatin (30 or 100 µM) or DMSO vehicle, measured every 24 h over 5 days. Two-way ANOVA (time and treatment as two independent factors) was applied to analyse the difference between each treatment and the DMSO control. Dunnett’s post hoc multiple comparison test was then performed to adjust the significance. The data displayed is mean ± SEM of triplicates of three independent experiments.
Ivacaftor suppressed the growth of 3D bioprinted cells in a dose-dependent manner
After being embedded in 3D bioprinted hydrogels, all of the four HGSOC cell lines showed a slight increase in resistance to ivacaftor, with IC50 values rising slightly, ranging from 11.85 to 18.19 µM (Figure 4).
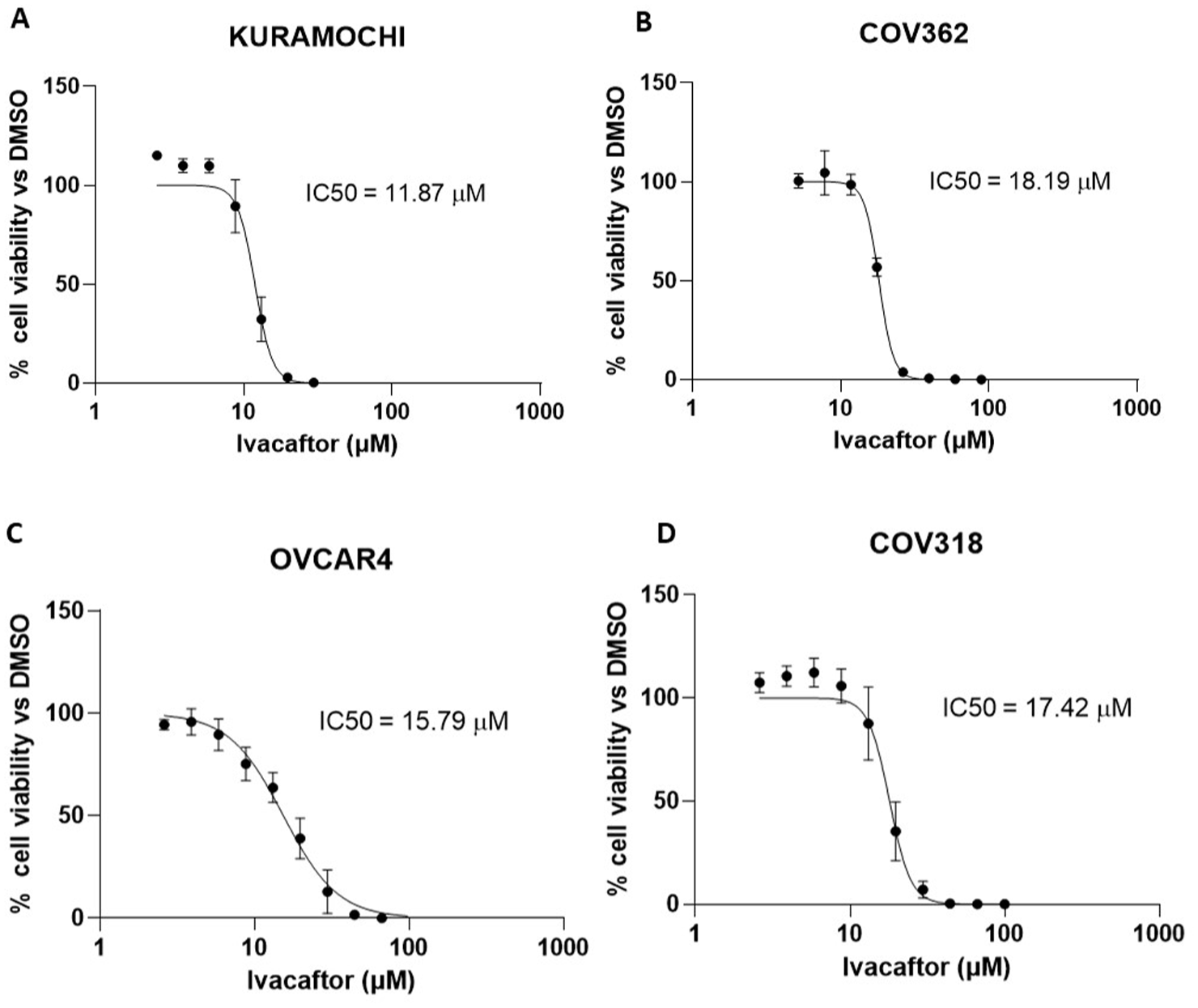
Bioprinted 3D culture models of HGSOC cell lines are sensitive to the cytotoxic effects of ivacaftor. Dose response curves for ivacaftor-treated 3D models of HGSOC cell lines (a) COV318, (b) COV362, (c) Kuramochi and (d) OVCAR4. Data represent mean cell viability ± SEM of n = 3 independent experiments, as a percentage of DMSO control versus concentration of ivacaftor (µM) plotted on a log10 scale. IC50 values were extrapolated from non-linear regression curve fitting performed in GraphPad Prism (GraphPad Software, USA).
Ivacaftor inhibited both HRD and HRP HGSOC organoid growth and induced apoptosis in a dose-dependent manner
ROR1 IHC (6D4) was performed in a clinical cohort of HGSOC patients from which patient-derived organoids were established. ROR1 high models OC029, OC043 and OC058 were selected for dose–response analysis (Figure S3). Clinicopathological characteristics of the patients were shown in Supplemental Table 2. The organoids exhibited a similar range of IC50 values as the 3D bioprinted cell lines, ranging from 11.18 to 14.11 µM. Both OC062 (HRP) and OC075 (HRD) organoids exhibited significantly higher Annexin V levels following 15 µM ivacaftor treatment compared to vehicle control (Figure 5(d)–(g)).

HGSOC patient-derived organoids are sensitive to ivacaftor. Dose–response curves for HGSOC organoids (a) OC029 (gBRCA1mut), (b) OC043 (gBRCA1mut) and (c) OC058 (BRCA wildtype, HRD). Data represents mean ± SEM, as a percentage of DMSO control concentration of ivacaftor (µM) plotted on a log10 scale. IC50 values were extrapolated from non-linear regression curve fitting performed in GraphPad Prism (GraphPad Software, USA). (d, e) Cells expressed Annexin V in patient-derived organoids (OC062 and OC075) treated with 15 µM ivacaftor or vehicle control at 24, 48 and 72 h measured using IncuCyte real-time cell imaging system. (f, g) Representative images were extracted from IncuCyte system at 72 h post-treatment. Scale bars represent 400 µm.**p < 0.01.
Ivacaftor did not induce DNA damage, DNA repair-mediated apoptosis or stall cell cycle in HGSOC cells
Flow cytometry was performed to assess induction of apoptosis by ivacaftor and the effects of ivacaftor treatment on cell cycle and DNA damage. Carboplatin was also used to treat the HGSOC cells, as a positive control, as it is known to exert its cytotoxic effects by causing DNA damage, cellular apoptosis and affecting cell cycle.47,48 Cell lines were treated with 15 µM ivacaftor or 30 µM carboplatin for 24, 48 or 72 h, and then immunostained with antibodies targeting cleaved PARP and phophorylated-H2AX (γ-H2AX) to detect apoptosis and DNA damage. Cells were also stained with DAPI to identify the distribution of cell cycle phases.
In COV362, 15 µM ivacaftor induced apoptosis compared to DMSO vehicle at 72 h (Figure 6(a)). There was a significant increase in cleaved PARP after 72 h of ivacaftor treatment (ratio = 1.521, p = 0.023), indicating an increase in the population of apoptotic cells. Treatment with 30 µM carboplatin also increased cleaved PARP compared to DMSO vehicle after 24 h (ratio = 1.231), 48 h (ratio = 1.501, p < 0.05) and 72 h (ratio = 1.949). Treatment with 15 µM ivacaftor did not cause an increase in DNA damage in COV362 (Figure 6(c)), measured by γ-H2AX, compared to DMSO vehicle after 24 h (ratio = 1.073), and 72 h (ratio = 1.045), a small decrease in DNA damage was seen after 48 h (ratio = 0.853, p = 0.0303). As expected, treatment with 30 µM carboplatin increased γ-H2AX levels compared to DMSO vehicle after 24 h (ratio = 4.053), and significantly increased γ-H2AX levels after 48 h (ratio = 7.595, p = 0.0391) and 72 h (ratio = 6.479, p = 0.0370).

Ivacaftor induced cell apoptosis in HGSOC. Cell apoptosis (a, b), DNA damage (c, d) and cell cycle (e, f) analyses of COV362 and KURAMOCHI cell lines were treated with 15 µM ivacaftor, 30 µM carboplatin or DMSO vehicle and measured at 24, 48 and 72 h. One sample t test was used to compare the fluorescence intensity of each fluorescent label between treatments. The proportion of cells in cell cycle phases were compared using a one-way ANOVA with Tukey’s post hoc test for multiple comparisons. Data were presented as the mean ± SEM from three replicate experiments of each cell line. Significance was set at p < 0.05.*p < 0.05, **p < 0.01.
Treatment with 15 µM ivacaftor or 30 µM carboplatin did not significantly affect the proportion of cells in G1 phase (DMSO vehicle mean = 67.2%, ivacaftor mean = 68.3%, carboplatin mean = 69.4%), S phase (DMSO vehicle mean = 24.3%, ivacaftor mean = 22.3%, carboplatin mean = 24.2%) and G2 phase (DMSO vehicle mean = 8.5%, ivacaftor mean = 9.4%, carboplatin mean = 6.4%) of the cell cycle after 24 h compared to DMSO vehicle in COV362 (Figure 6(e)). After 48 h, there was a decrease in proportion of cells in G1 phase (DMSO vehicle mean = 64.9%, ivacaftor mean = 53%, carboplatin mean = 44%), an increased in proportion of cells in S phase (DMSO vehicle mean = 21.3%, ivacaftor mean = 29.5%, carboplatin mean = 36%) and no difference in the G2 phase (DMSO vehicle mean = 13.8%, ivacaftor mean = 17.5%, carboplatin mean = 20%) compared to DMSO vehicle when treated with 15 µM ivacaftor or 30 µM carboplatin. The decrease in the proportion of cells in G1 phase continued to 72 h (DMSO vehicle mean = 59.2%, ivacaftor mean = 55.5%, carboplatin mean = 28.5%), along with the increase in the proportion of cells in S phase (DMSO vehicle mean = 25.9%, ivacaftor mean = 33.2%, carboplatin mean = 51%; p < 0.01) and no difference in proportion of cells in G2 phase (DMSO vehicle mean = 14.9%, ivacaftor mean = 11.3%, carboplatin mean = 20.5%) compared to DMSO vehicle after 72 h when treated with 15 µM ivacaftor or 30 µM carboplatin. As expected, treatment with 30 µM carboplatin for 72 h decreased the proportion of cells in the G1 phase approaching significance, and significantly increased the proportion of cells in the S phase compared to DMSO vehicle.
KURAMOCHI did not show any increase in cleaved PARP (24-h ratio = 0.786, p = 0.015; 48-h ratio = 0.802; 72-h ratio = 0.912) and γ-H2AX (24-h ratio = 0.914; 48 h ratio = 0.975; 72 h ratio = 1.074) compared to DMSO vehicle when treated with 15 µM of ivacaftor (Figure 6(b) and (d)). However, treatment with 30 µM carboplatin increased apoptosis in KURAMOCHI compared to DMSO vehicle and ivacaftor treatment after 24 h (ratio = 1.139, p = 0.0036), 48 h (ratio = 1.909) and 72 h (ratio = 1.791, p = 0.0329), and significantly increased DNA damage compared to DMSO vehicle after 24 h (ratio = 3.62, p = 0.029), 48 h (ratio = 11.5, p = 0.008) and 72 h (ratio = 17.18, p = 0.035). There was no significant difference in the proportion of cells in the G1 phase (DMSO vehicle mean = 46%, ivacaftor mean = 44.5%, carboplatin mean = 41.9%), S phase (DMSO vehicle mean = 42.8%, ivacaftor mean = 42.2%, carboplatin mean = 46.7%) and G2 phase (DMSO vehicle mean = 11.2%, ivacaftor mean = 13.3%, carboplatin mean = 11.4%) when treated with DMSO vehicle, 15 µM ivacaftor and 30 µM carboplatin for 24 h (Figure 6(f)). After 48 h of treatment, there was no significant difference in the proportion of cells in the G1 phase (DMSO vehicle mean = 43.9%, ivacaftor mean = 44.3%, carboplatin mean = 43.7%), S phase (DMSO vehicle mean = 44%, ivacaftor mean = 40.6%, carboplatin mean = 43.4%) and G2 phase (DMSO vehicle mean = 12.1%, ivacaftor mean = 15.1%, carboplatin mean = 12.9%). There is no significant difference in proportion of cells in the S phase (DMSO vehicle mean = 34.6%, ivacaftor mean = 35.8%, carboplatin mean = 42.2%) after treatment with DMSO vehicle, 15 µM ivacaftor or 30 µM carboplatin. However, there was a non-significant decrease in proportion of cells in the G1 phase (DMSO vehicle mean = 51.1%, ivacaftor mean = 40.8%, carboplatin mean = 32.1%) and a non-significant increase in proportion of cells in the G2 phase (DMSO vehicle mean = 14.3%, ivacaftor mean = 23.4%, carboplatin mean = 25.7%) after 72 h treatment with 15 µM ivacaftor and 30 µM carboplatin compared to DMSO vehicle.
Ivacaftor induced downstream regulation of ROR1 signalling in HGSOC cell lines
Immunoblotting was conducted to assess the expression of proteins involved in downstream pathways previously shown to be regulated by ROR1 inhibition, following treatment with 15 µM ivacaftor for 48 h (Figure 7(a)). Two HGSOC cell lines, KURAMOCHI and COV362, with high ROR1 expression, along with their corresponding ROR1 knockout derivatives, were analysed. BMI-1 was selected as a key regulator of cancer stem cell self-renewal; pAKT and AKT were included to assess AKT phosphorylation in the PI3K/AKT pathway and cleaved PARP and cleaved Caspase 3 were used as markers of DNA damage. GAPDH was used as a reference control.

Western blot and corresponding densitometry analyses of key regulators involved in ROR1-associated downstream signalling pathways following 48 h 15 µM ivacaftor treatment in HGSOC cell lines and their ROR1 knockout lines. (a) Blots are representatives of three independent experiments. The corresponding raw blots for each experiment are provided in Figure S4. (b–g) Densitometry analysis of protein of interest was performed using Image J and normalised against the loading control (GAPDH) level. Data represent as mean ± SEM.
The upper band (around 130 kDa) of ROR1 showed a moderate deduction following ivacaftor treatment compared to the vehicle control in COV362 only (p = 0.07, Figure 7(b)), indicating that as expected for an allosteric modulator, ivacaftor does not directly reduce expression of ROR1. In the KURAMOCHI wild-type (WT) cell line treatment with ivacaftor showed decreases in downstream signalling markers pAKT (p = 0.01) and BMI-1 (p = NS, Figure 7(a), (d) and (e)) and increased apoptosis markers cPARP (p = NS) and cCas3 (p = 0.03; Figure 7(a), (f) and (g)) when compared to vehicle controls, indicating the allosteric binding of ivacaftor elicits an effect on ROR1 downstream signalling. KURAMOCHI ROR1 KO cell lines showed a very similar trend for ivacaftor treatment compared to vehicle control, but the trends did not reach statistical significance. These results suggest that the downstream signalling response to ivacaftor is not mediated exclusively through ROR1 expression. Interestingly, it was observed that ROR2 expression was present in both KURAMOCHI WT and ROR1 KO cell lines (Figure 7(a) and (c)) indicating the downstream response may be mediated through ROR2.
A second cell line with high expression of ROR1, COV362 was also used to investigate the induction of downstream regulation of ROR1 signalling with ivacaftor treatment. Similar to KURACMOCHI, COV362 WT expressed both ROR1 and ROR2 (Figure 7(a) and (c)). COV362 WT treatment with ivacaftor showed significant decreases in downstream signalling markers pAKT (p = 0.04) and BMI-1 (p = 0.05; Figure 7(a), (d) and (e)) and significant increases in apoptosis markers cPARP (p = 0.04) and cCas3 (p = 0.01; Figure 7(a), (f) and (g)) when compared to vehicle controls. However, in the COV362 ROR1 KO cell line, there was no expression of ROR1 or ROR2 (Figure 7(a) and (c)). In the absence of ROR1 and ROR2 expression, downstream signalling markers pAKT and BMI-1 were not significantly different with ivacaftor treatment when compared to vehicle control (Figure 7(a), (d) and (e)) and apoptosis markers cPARP and cCas3 were expressed at very low levels with no difference between vehicle and ivacaftor treatment (Figure 7(a), (f) and (g)).
Taken together, these results suggest that ivacaftor modulates downstream signalling in ROR1-expressing models and induces apoptotic responses by acting as an allosteric modulator that potentially involves both ROR1 and ROR2. 49 ROR1 knockout-dependent differences are most evident in COV362 cells, while residual effects observed in KURAMOCHI cells may indicate compensatory mechanisms. ROR2 appears to be playing a compensatory role in the absence of ROR1, as the downstream ROR1 signalling response to ivacaftor was not observed when both ROR1 and ROR2 were not expressed.
Discussion
In the absence of early detection, effective therapy is urgently needed for highly aggressive and treatment-resistant HGSOC. While efforts towards drug discovery have been made, they have been significantly hindered by the time and cost required for approval. Traditional drug development typically takes approximately 12–16 years and costs over 1 billion US dollars. 50 In contrast, drug repurposing greatly reduces the time and effort by leveraging existing knowledge of clinically available drugs, bypassing most preclinical and early clinical trial phases.51–54 As a result, repurposing a drug takes an average of 6.5 years and costs approximately 300 million US dollars.55,56 We applied a systematic drug repurposing approach to streamline the translation of novel targeted therapies for HGSOC. The drugs were filtered after integrating data from comprehensive pharmacology and drug screening databases as well as HGSOC-specific preclinical models for in vitro testing, which holds great potential for progressing to early phase clinical trials with existing in vivo or phase I safety profiles.
Through our in silico and in vitro screening, ivacaftor (also known as Kalydeco or VX-770), approved for the treatment of cystic fibrosis 57 emerged as a candidate for repurposing in HGSOC. Ivacaftor, an allosteric modulator, binds to alternate sites to canonical receptor ligands, with the aim of altering the response to endogenous ligand binding. In 2006, Van Goor et al. 58 identified allosteric modulator compounds with cystic fibrosis transmembrane conductance regulator (CFTR) potentiator activity by conducting high-throughput screening of 228,000 compounds. The most promising lead compound from the screen was VRT-484, which required further medicinal chemistry optimisation to VRT-715, followed by further structural modifications to increase solubility, ultimately leading to final allosteric modulator compound VX-770, which would become known as ivacaftor. VX-770 was not designed to specifically bind to CFTR; therefore, it is highly likely to bind to allosteric sites in common with other receptors.
In 2012, ivacaftor was the first CFTR potentiator approved for patients aged ⩾6 years with CFTRG551D mutation.59,60 At the time of FDA approval, ivacaftor was known to be an allosteric modulator but the exact mechanism of action was still unknown. From 2012 to 2023 extensive research was conducted into elucidating the mechanism of action of ivacaftor, with the final publications in 2023 describing its effect as an allosteric modulator for CFTR, primarily by increasing the probability that the chloride channel remains open, thereby enhancing CFTR gating activity.43,44
Following 150 mg twice daily dosing of ivacaftor, the adult dose for CF monotherapy or in multidrug regimens, the average plasma concentrations at steady state (Cssav) across four studies in healthy individuals ranged from 0.79 to 1.5 µg/mL, and Cmax at steady state ranged from 1.16 to 1.97 µg/mL (2.01–3.82 and 2.95–5.0 µM, respectively), although considerable interpatient variability was observed (%CV for these parameters ranging between approximately 30% and 70%). 61 With the exception of respiratory tract reactions, adverse events at the standard dose regimen are infrequent (small difference from placebo) or mild in severity. Respiratory tract reactions occurred at a frequency of 13.3% in ivacaftor-treated CF patients following 48 weeks of treatment (63.3% vs 50.0% on placebo). 62 Higher doses were evaluated in early phase trials, including 450 mg twice daily for 5 days in healthy subjects: Cssav and Cmax were 4.3 µg/mL (11.0 µM) and 5.5 µg/mL (14.01 µM), respectively. 63 Ivacaftor pharmacokinetics and adverse events would need to be assessed in the HGSOC population, especially in older people not represented in CF trials, although the data from such trials suggest a generally favourable safety profile.
The HGSOC cell lines selected for in vitro analysis were based on positivity for ROR1, with variability in their BRCA genotypes. OVCAR4 and COV318 are BRCA WT while KURAMOCHI and COV362 carry BRCA1/2 mutations. 34 Despite harbouring BRCA1 mutations, COV362 expresses an alternative isoform of BRCA1 protein which is functionally classified as HRP. 64 The efficacy of single ivacaftor treatment observed in the BRCA WT or HRP HGSOC cell lines and patient-derived organoids (OC058 and OC062) is encouraging, as this subgroup have shown limited benefits from standard maintenance therapies involving chemotherapy and the antiangiogenic agent bevacizumab, or PARPi. 4 In application for the FDA approval of ivacaftor, two 2-year carcinogenicity studies conducted in rats and mice found that there were no drug-related neoplasms. This may suggest that there is no significant DNA damage related to ivacaftor use, which is consistent with the results, and ivacaftor does not act as a DNA-damaging agent. 63 Ivacaftor was shown to induce cellular apoptosis in glioblastoma stem cells, using markers of both early and late apoptosis. 65 In this study, COV362 cell lines exhibited an increase in apoptotic markers (e.g., cleaved PARP or cleaved Caspase 3; Figures 6 and 7), as well as increased cell apoptosis (Figure 3(f)) following ivacaftor treatment. Treatment with 15 µM ivacaftor significantly increased apoptosis compared to vehicle control up to 72 h when detected with the early apoptotic marker Annexin V, but not significantly in KURAMOCHI. When apoptosis was assessed using cleaved PARP and Caspase 3 (markers of late apoptosis), KURAMOCHI and COV362 cells showed increased apoptotic activity by Western blot but not by flow cytometry. KURAMOCHI is a BRCA2-mutant cell line, and genetic inactivation of BRCA2 in cancer cell lines has been shown to increase TRAIL-R-mediated apoptosis, 66 suggesting that measurement of Annexin V and cleaved PARP may not represent the apoptotic processes in the KURAMOCHI cell line. Taken together, this suggests a mechanism of cell apoptosis, independent of DNA damage upon ivacaftor treatment. A previous study on the off-target effects of ivacaftor (not related to CFTR) found that ivacaftor induced oxidative stress by increasing reactive oxygen species (ROS) levels in CFTR-deficient cells. 67 However, the study did not identify which proteins ivacaftor bound to that triggered the ROS increase. One possible mechanism could involve PI3K/AKT inhibition following ivacaftor treatment as shown in our study. Ivacaftor inhibited ROR1 signalling, as evidenced by the significant reduction of pAKT/AKT and BMI-1 in COV362 WT but not in the COV362 ROR1 knockout, suggesting that its effects are at least partly mediated through ROR1. Interestingly, ROR1 knockout KURAMOCHI cells still showed a trend towards reduced signalling upon ivacaftor treatment. We hypothesise that this may be due to compensatory activity from ROR2, which remains expressed after ROR1 deletion in the KURAMOCHI cell line. This trend was not seen in the absence of both ROR1 and ROR2 in the COV362 ROR1 KO cell line. Given that ROR1 signalling is initiated by Wnt5a binding to a ROR1–ROR2 dimer, 16 the presence of ROR2 alone appears to be sufficient to activate downstream signalling cascades.
Chemoresistance has become a key contributor to the poor prognosis observed in HGSOC. We proposed a therapeutic approach targeting ROR1 to address this issue. ROR1 has been correlated with cancer cell stemness and chemoresistance via various pathways including BMI-1.6,19 In breast cancer, ROR1 regulates the drug efflux pump ABCB1, 15 which has been identified as a key mechanism contributing to chemoresistance in ovarian cancer. 68 The PI3K/AKT pathway acts as a central hub in multidrug resistance, regulating downstream processes that ultimately enhance tumour cell survival after drug treatment (reviewed in Liu et al. 69 ). Ivacaftor, a known allosteric receptor modulator, triggered inhibition of the PI3K/AKT pathway and reduced BMI-1 level in HGSOC cell lines. This suggests ivacaftor shows promise for the treatment of chemoresistant diseases. Future studies combining ivacaftor with standard of care chemotherapies for the treatment of chemoresistant HGSOC should be explored.
Despite the promising outcomes observed in this study, several limitations should be acknowledged. Although previous studies have reported various post-translational modifications of ROR1 relevant to its functional signalling, antibodies that specifically detect the activated phosphorylated form of ROR1 (p-ROR1) remain limited and insufficiently validated. We assessed the only commercially available p-ROR1 antibody and were unable to obtain reliable results which presented limitations on our ability to confirm the direct inhibition of ROR1 activity by ivacaftor. In addition, we were unable to draw firmer conclusions from a small-molecule inhibitor class control for ROR1. Finally, we did not assess toxicity in non-cancerous control cells in this study. For context, key advantages of the drug repurposing approach are the availability of published safety data from preclinical studies, early-phase clinical trials and post-market surveillance of clinical use of the drug. Ivacaftor has demonstrated a favourable toxicity and safety profile in all of these settings. Nevertheless, additional evaluation of the limitations described is recommended for further validation of ivacaftor as a potential repurposed drug for HGSOC.
Conclusion
Ivacaftor demonstrated significant single agent anti-tumour potential in preclinical HGSOC models, supporting its further investigation as a repurposed therapy for ROR1-expressing HGSOC.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251409010 – Supplemental material for Repurposing of ivacaftor shows potential to treat ROR1 expressing high-grade serous ovarian cancer
Supplemental material, sj-docx-1-tam-10.1177_17588359251409010 for Repurposing of ivacaftor shows potential to treat ROR1 expressing high-grade serous ovarian cancer by Dongli Liu, Michelle Wong-Brown, Farhana Amy Sarker, Bayley Matthews, Jessica Morrison, Kristie-Ann Dickson, Amani Alghalayini, Jana Stojanova, King Man Wan, Jennifer Duggan, Christine Loo, Gill Stannard, Elyse Powell, Brigitte Hodder, Ellen Barlow, Deborah J. Marsh, Caroline E. Ford and Nikola A. Bowden in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We gratefully acknowledge the patients who generously donated biospecimens for this research. We appreciate the valuable contributions of our consumer working group (led by co-author Gill Stannard and including Jacinta Frawley, Kristin Young, Amanda Warrington and Melissa Tooney) in the drug shortlisting process.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.