
Abstract
Non-clear cell renal cell carcinomas (nccRCC) comprise a heterogeneous group of rare malignancies, accounting for approximately 20% of all kidney cancers. Given the rarity of these diverse subsets of RCC, the treatment paradigm for nccRCC is often based on treatment strategies utilized in the management of clear cell renal cell carcinoma (ccRCC), which may not fully address the distinct molecular and genetic drivers unique to nccRCC tumors. However, recent advances in the molecular characterization of nccRCC have led to the identification of new therapeutic targets, resulting in the development of more targeted therapies for nccRCC treatment. Furthermore, the role of molecular characterization of renal tumors is emphasized in the 2022 World Health Organization reclassification of genitourinary tumors, given that delineation of renal tumor subtypes now also relies on genetic markers. In order to highlight this evolving treatment landscape, our review provides a comprehensive summary of recent progress related to the biology and management of nccRCC subtypes.
Plain language summary
Non-clear cell renal cell carcinomas are rare types of kidney cancer that account for about 20% of all cases. These cancers are very diverse and often treated like the more common clear cell type, even though their biology and genetics are different. In 2022, the World Health Organization updated the classification of these rare kidney cancers, making it easier to identify specific subtypes and develop targeted treatments. This review highlights recent advancements in understanding biology and explores new treatment strategies that could help patients with these rare kidney tumors.
Introduction
Renal cell carcinoma (RCC) accounts for approximately 4%–5% of all cancers in the United States with an estimated 81,610 new cases estimated in 2024. 1 Clear cell RCC (ccRCC) accounts for 80% of all RCC cases, while the remaining 20% of cases include non-clear cell RCC (nccRCC). 1 In 2022, the World Health Organization (WHO) reclassified nccRCC into categories that better reflect the distinct genetic, molecular, and histological features of nccRCC. 2 This reclassification increasingly relies on molecular and genetic markers for categorization. The movement toward molecular classification allows for the incorporation of tumor genetics and biomarkers into study design when planning future nccRCC trials, an approach that implements a shift toward precision medicine. 3
The 2022 WHO classification system for nccRCC introduced significant updates, including the recategorization of existing tumor types and the addition of new entities. Major recategorizations include papillary RCC no longer being divided into type 1 and type 2 based on histological and characteristic genetic alterations. Former type 1 RCC accounts for the entirety of the papillary RCC category, while the genetically and morphologically diverse former type 2 has been separated into distinct molecularly defined categories. 2 Novel molecularly defined categories such as fumarate hydratase-deficient RCC (fhRCC), anaplastic lymphoma kinase-rearranged RCC, TFE3-rearranged RCC and TFEB-altered RCC (formerly microphthalmia-associated transcription factor (MiT) family of RCCs), and ELOC (formerly TCEB1) mutated RCC.2,4 SMARCB1 (INI1)-deficient renal medullary carcinoma (RMC) replaced RMC classification in the 2022 classification as the subtype characterized by loss of nuclear expression of SMARCB1.2,5 The classification of oncocytic and chromophobe renal tumors was broadened to include “oncocytic renal neoplasms of low malignant potential NOS,” which describes low-grade oncocytic tumors or eosinophilic tumors that do not fit into other categories. 2 Another change includes the recategorization of clear cell papillary RCC to clear cell papillary renal cell tumor, reflecting the less aggressive nature of the disease. 2
Current treatment options focus on immunotherapy, vascular endothelial growth factor (VEGF) inhibitors, and MET inhibitors, which, in part, have been adapted from ccRCC data.6–9 Due to the molecular, genetic, and pathological diversity of nccRCC, as well as their comparative rarity compared to ccRCC, randomized clinical trials are challenging but necessary to advance nccRCC outcomes. This review aims to describe the advancements in biology and treatment strategies for nccRCC.
Papillary renal cell carcinoma
Papillary renal cell carcinoma (pRCC), historically type 1 pRCC, accounts for 10%–15% of all RCCs. 10 The incidence is twofold higher in men, with a median age of diagnosis around 75 years. 11 Although possible contributors to the development of pRCC have been identified, its etiology remains unclear.
Studies that have evaluated the genetic and molecular profile of the pRCC have demonstrated a variety of chromosome gains in pRCC.12,13 The Cancer Genome Atlas Research Network (TCGA) study demonstrated nearly universal chromosome 7 and 17 gains in the type 1 papillary RCC predominant subgroup of patients. Chromosome 2, 3, 12, 16, and 20 gains were also observed, but at a lesser frequency. 14 MET is an oncogene located on chromosome 7q21-q31, which encodes the tyrosine kinase receptor (RTK) for the hepatocyte growth factor. 15 Phosphorylation of RTK triggers a cascade of downstream signals, such as PI3K, MAPK, STAT3, and Src. 16 The PI3K pathway further activates Akt and mTOR, which promote cell growth and suppress apoptosis. The MAPK pathway leads to cellular proliferation and differentiation. STAT3 activation ultimately promotes cell proliferation and transformation. 17 The activation of the Src pathway then results in NF-κB activation, which also promotes cell proliferation and transformation. 18
MET mutations have been identified in multiple solid tumors. The TCGA study, which investigated the molecular characterization of pRCC, demonstrated that 17% of the type 1 papillary RCC tumors had MET mutations. 14 Similarly, Schmidt et al. 19 reported MET mutations in 17 of 129 (13%) patients without any family history of pRCC. In another report, Schmidt et al. found germline mutations in four out of seven patients with hereditary papillary renal cell carcinoma. In the same report, Schmidt et al. 20 reported that 3 of 60 (5%) patients had somatic mutations in the MET gene. In contrast, Albiges et al. 21 reported copy number alterations in the MET gene in 40 of 47 (81%) patients with type 1 pRCC. Multiple clinical trials have recently evaluated the role of MET inhibition as a therapeutic target in the context of pRCC; for instance, savolitinib, a selective tyrosine kinase inhibitor (TKI) for MET, has been investigated in MET-driven pRCC patients.
Chromosome Y loss is another genetic feature identified in pRCC22,23; however, its implications for clinical practice are unclear. Büscheck et al. reported chromosome Y loss in 84% of the patients with type 1 pRCC (n = 120). In a subgroup analysis evaluating recurrence-free survival in pRCC, the Y loss group had significantly longer recurrence-free survival. However, multivariate analysis assessing variables such as tumor grade, stage, and chromosome Y status demonstrated that chromosome Y status had no prognostic significance for clinical outcomes. 24
Neurofibromin2 (NF2) tumor-suppressor gene alterations (GAs) have also been observed in papillary RCC patients. The TCGA study demonstrated that mutations on the Hippo signaling pathway, which includes NF2, were present in 3% of type 1 pRCC patients. 14 Hacking et al. analyzed 362 patients with pRCC and found that 43 (12%) of patients had NF2 alterations, suggesting that NF2 may be a potential target for treatment. They also identified co-occurring mutations, including CDKN2A, SETD2, and BAP1, as well as higher PD-L1 expression in NF2-mutant tumors, suggesting potential sensitivity to immune checkpoint inhibitors (ICIs). 25
Further investigation into NF2 alterations aimed to identify a potential subgroup of renal tumors driven by this genetic alteration. Argani et al. reported a case series of eight patients, describing a previously unrecognized RCC associated with NF2 GAs. In their report, seven out of eight tumors exhibited papillary architecture, and they characterized these tumors as biphasic hyalinizing psammomatous RCC. The authors suggested that NF2 GAs can be associated with a distinct type of RCC and pointed to the potential for targeted therapies for this specific driver. 26 In support of their findings, another study analyzed 414 clinically advanced pRCCs and confirmed the presence of NF2 GAs in 12% of cases, which were exclusively seen in stage IV pRCC, highlighting the role of NF2 GAs in driving more aggressive forms of RCC. Given that inactivation of the NF2 gene leads to the loss of a tumor suppressor protein, Merlin, that plays a pertinent role in multiple signaling pathways including mTOR, there may be a putative role for mTOR inhibitors in treating pRCC tumors with underlying NF2 GAs. 27 The genetic changes observed in nccRCC are summarized in Figure 1.
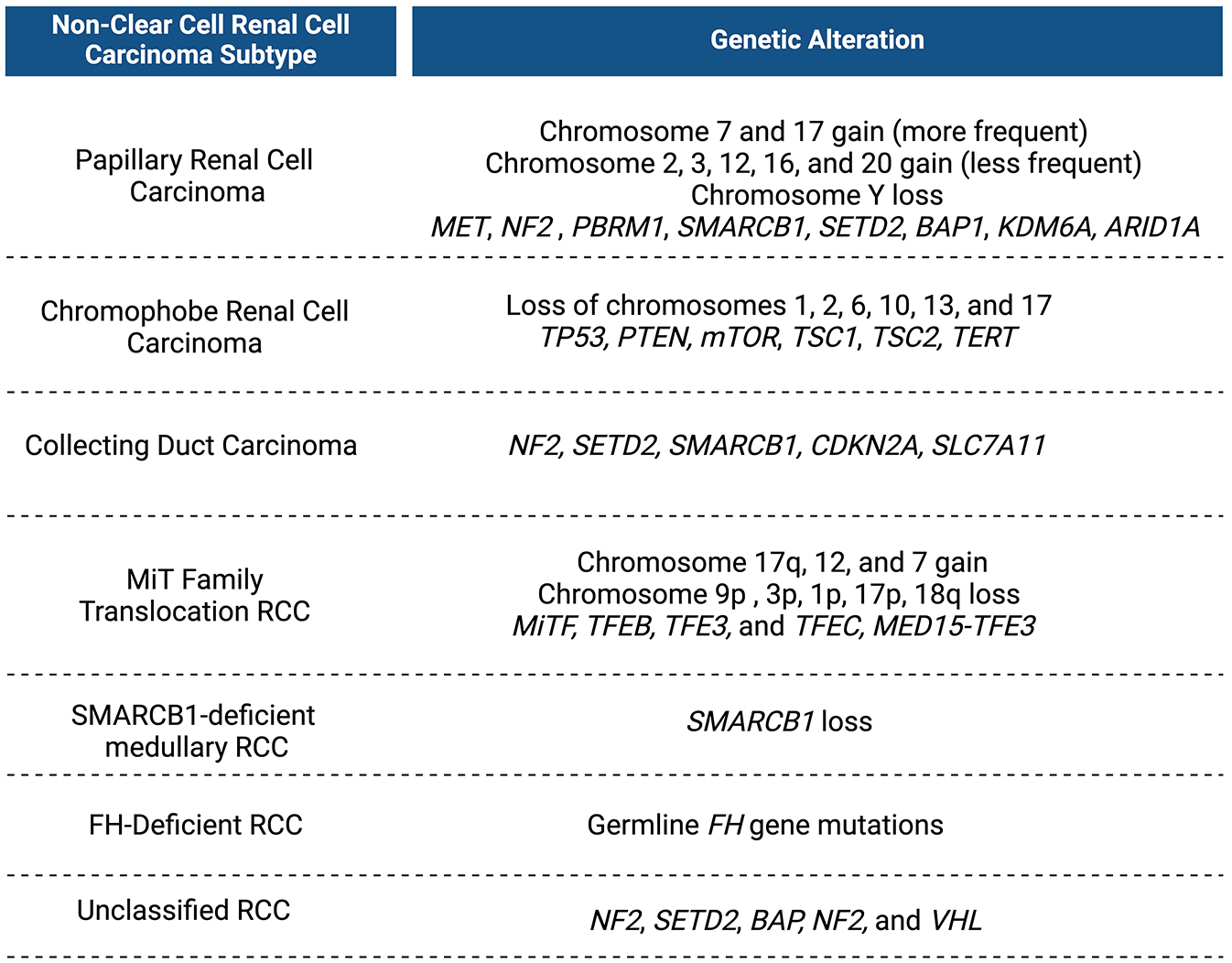
A summary of genetic alterations observed in non-clear cell renal cell carcinoma by subtype.
Chromatin remodeling complexes are well known for their relationship with cancer. The SWItch/Sucrose Non-Fermentable (SWI/SNF) family of chromatin remodeling complexes regulates nucleosome positioning. SWI/SNF complexes have multiple roles in regulating cell differentiation and transcriptional activity.28,29 SWI/SNF mutations, which include PBRM1 and SMARCB1, are present in nearly 25% of all cancers.30,31 SWI/SNF family and other mutations, most significantly SETD2, BAP1, and KDM6A, in chromatin remodeling complexes were found in 20% of type 1 pRCC patients. 14 Mutations in the SWI/SNF complex and their impact on tumor cells are summarized in Figure 2. Next-generation sequencing (NGS) analysis from the phase II trial of bevacizumab plus everolimus in advanced nccRCC revealed that a mutation in ARID1A, a member of the SWI/SNF complex, might be correlated with longer progression-free survival (PFS). In that report, 14 patients were identified with papillary features. Five patients carrying the ARID1A mutation had PFS longer than 6 months, and nine patients without the mutation had PFS shorter than 6 months. 32

Mutations in the SWI/SNF chromatin remodeling complex and their impact on tumor cells.
Management
Despite clear biological differences, treatment recommendations for pRCC historically mirrored ccRCC due to the paucity of prospective trial data. This has led to the development of several phase II trials, either stand-alone or of variant histology, serving as the framework for current pRCC treatment guidelines. The advent of SUPAP and RAPTOR, single-arm phase II trials, introduced sunitinib (VEGF inhibitor) and everolimus (mTOR inhibitor) as viable first-line therapies in pRCC.33,34 However, the subsequent completion of the ASPEN and ESPN trials established sunitinib as the standard of care option. The ASPEN trial compared sunitinib to everolimus in nccRCC patients, with sunitinib demonstrating median progression-free survival (mPFS) benefit of 8.3 months (80% confidence interval (CI) 5.8–11.4) vs 5.6 months (80% CI 5.5–6.0) for everolimus (hazard ratio (HR) 1.41 (80% CI 1.03–1.92); p = 0.16). 35 Additionally, in pRCC patients, ASPEN noted an objective response rate (ORR) of 24% versus 5% in sunitinib and everolimus groups, respectively. The ESPN trial data did not recognize a similar benefit and reported comparable PFS and OS between groups. 36
Current understanding of germline and sporadic MET alterations in pRCC led to a series of trials investigating MET inhibition. The first was the prospective phase II study of foretinib, an oral multikinase inhibitor targeting c-MET (hepatocyte growth factor receptor) and vascular endothelial growth factor receptor (VEGFR), in 74 patients with advanced pRCC. Though the trial failed to reach its predefined response rate of 25% (13.5%, 95% CI 6.7–23.0), the reported PFS of 9 months compared favorably with prior experience utilizing VEGF and mTOR inhibitors. 37 Of note, germline MET alterations were associated with improved response to foretinib compared to non-MET-mutated patients. Phase II data showed promising activity with savolitinib, a selective MET inhibitor, within MET-driven pRCC, justifying the subsequent development of the phase III trial, SAVOIR. 38 SAVOIR, allocated 60 patients with MET-driven advanced pRCC to savolitinib versus sunitinib. 39 While the study was terminated early due to recruitment challenges, savolitinib demonstrated a longer mPFS compared to sunitinib (7.0 months (95% CI, 2.8–not calculated) vs 5.6 months (95% CI, 4.1–6.9); HR 0.71 (95% CI, 0.4–1.4); p = 0.31) and a higher ORR (27% vs 7%), although the difference was not statistically significant. More recently, PAPMET, a phase II randomized trial, compared sunitinib to three MET kinase inhibitors (cabozantinib, crizotinib, and savolitinib) in 147 advanced pRCC patients. 40 Cabozantinib was the most efficacious compared to sunitinib with a median PFS of 9 versus 5.6 months (HR 0.60; 95% CI 0.37–0.97, p = 0.019) and ORR 23% versus 4% (p = 0.010), respectively. Neither savolitinib nor crizotinib improved outcomes compared with sunitinib. The PAPMET trial was the first study to show clinical and statistical benefit over an established standard of care within pRCC, establishing cabozantinib as the preferred option.
More recently, the introduction of immunotherapy within RCC management has led to the creation of several monotherapy and combination trials examining the efficacy of ICIs in nccRCC. Checkmate 374, an open-label, phase IIIb/IV study evaluating nivolumab monotherapy, was the first to exhibit the promising efficacy of ICIs in patients with advanced nccRCC. In the nccRCC cohort (44 patients), 24 patients confirmed papillary histology with partial response and stable disease achieved in 8% and 37.5%, respectively. 41 To date, KEYNOTE-427 is the most extensive study examining the efficacy of PD-1 inhibitor pembrolizumab in pRCC, including 118 patients. The pRCC cohort showed promising antitumor activity with an ORR of 28%, a disease control rate of 47.5%, and an mPFS of 5.5 months. 42 Of note, within the nccRCC cohort, the ORR for pRCC was among the highest observed. In contrast, preliminary data from the AcSe trial, a nonrandomized multicenter trial evaluating nivolumab monotherapy in nccRCC, were discouraging for pRCC given that 29 pRCC patients (type 1: 1, type 2: 20, unclassified: 8) were included, with only 2 patients achieving partial response. 43 The promising antitumor activity of nivolumab plus ipilimumab in nccRCC gleaned from limited retrospective data led to the execution of the CheckMate 920 trial. Of the 18 pRCC patients, 28% exhibited responses with 1 complete response and 4 partial responses. 44
The rather promising use of ICI monotherapy in advanced nccRCC led to the evaluation of ICI combination with other approved targeted agents. The first being COSMIC-021 assessing atezolizumab plus cabozantinib in nccRCC with an ORR of 47% in pRCC patients. 45 Lee et al. 46 conducted a single-center phase II study of cabozantinib plus nivolumab, including 32 pRCC patients with an ORR of 47%. CALYPSO combined savolitinib with the PD-L1 antagonist durvalumab, enrolling 41 pRCC patients, of which 17 were MET-driven. For the entire cohort, the ORR was 29%, but in MET-driven patients, the ORR increased to 53%. 47 The results of CALYPSO led to the development of SAMETA, an ongoing phase III trial randomizing untreated pRCC patients to savolitinib + durvalumab versus durvalumab versus sunitinib. 48 KEYNOTE-B61, a single-arm, multicenter phase II trial evaluating lenvatinib and pembrolizumab in patients with previously untreated nccRCC, has recently shown the most promising antitumor activity within pRCC. A total of 93 (59%) pRCC patients were enrolled, showing an ORR of 54% and a disease control rate of 85%. Of the 93 patients, 10 had complete responses, 40 had partial responses, and 29 had stable disease. 49 As a result of the durable antitumor activity observed with ICI combination, there has been significant growth of exploration within this subset of clinical trials. For example, SWOG S2200 (PAPMET2) trial, a phase II randomized trial, is an ongoing study examining the role of cabozantinib and atezolizumab in pRCC. 50 Other unique combination trials based on promising ccRCC data are currently ongoing, including tivozanib + nivolumab (FORTUNE, NCT06053658) and zanzalintinib + nivolumab (STELLAR-304, NCT05678673).
Chromophobe renal cell carcinoma
Chromophobe renal cell carcinoma (chRCC) is the third most common subtype of RCC and accounts for ~5% of RCC cases. Based on the light microscopic appearance of the cells, chRCC has two variants: classic chRCC and eosinophilic variant chRCC. Studies suggested no difference in prognosis between classic and eosinophilic variants.51,52 In contrast, sarcomatoid differentiation is associated with a worse prognosis.53,54
Genomic profiling of chRCC differs from other RCC subtypes; chRCC has approximately threefold fewer somatic mutations compared to ccRCC. TCGA study only identified two significant mutations in chRCC: TP53, which was mutated in 32% of cases and is associated with reduced expression of p53 transcriptional targets, and PTEN, reported in 9% of cases. 55 Casuscelli et al. 56 explored the clinical significance of these mutations and found that metastatic disease had higher rates of TP53 and PTEN in metastatic disease. TCGA study reported that the loss of chromosomes 1, 2, 6, 10, 13, and 17 was seen in 86% of the cases. TP53 and PTEN are located on chromosomes 17 and 10. Roldan-Romero et al. reported that 17% of the cases had GAs in the mTOR pathway (mTOR, TSC1, and TSC2). They also noted that mTOR mutation was associated with a worse prognosis. However, mTOR inhibitors are not the preferred treatment agents for chRCC. Two studies found that mutations in the mTOR pathway genes confer sensitivity to mTOR inhibitors in RCC; however, those studies were mainly conducted on ccRCC and may not reflect sensitivity in chRCC.57,58
Recent advancements in understanding the molecular mechanisms driving dedifferentiation and aggressiveness in chRCC have highlighted its progression into more aggressive forms, such as sarcomatoid, anaplastic, and glandular dedifferentiation. Kapur et al. conducted a multiregion genomic analysis, identifying TP53 and PTEN mutations as key initiators of this process. These mutations often become homozygous following whole-genome duplication. This event, along with mTORC1 activation, contributes to increased tumor aggressiveness and metastatic potential. Interestingly, dedifferentiated chRCC shares molecular features with more aggressive RCC subtypes like ccRCC. These findings suggest that dedifferentiation plays a critical role in tumor progression and could inform future therapeutic approaches, including the potential for targeted treatments such as mTOR inhibitors or immune checkpoint inhibitors. 59
Telomerase reverse transcriptase (TERT) expression has gained attention as a diagnostic and prognostic marker in various cancers, including chRCC. TERT is closely linked to telomerase activity and the maintenance of telomere length, playing a key role in tumor development by ensuring chromosomal stability. 60 An analysis by TCGA revealed recurrent DNA rearrangement breakpoints within the TERT promoter region in 10% of chRCC cases, highlighting a potential mechanism for increased TERT expression and its association with tumor development. 55 Despite these findings, the exact role of TERT in driving chRCC progression remains underexplored, and its clinical utility as a biomarker is still under investigation. Future studies are needed to determine whether TERT expression could serve as a therapeutic target or a reliable prognostic indicator in chRCC.
Management
Currently, there are no dedicated prospective clinical trials providing insight into the optimal clinical management of chRCC. The existing approach, including anti-VEGF TKIs, mTOR inhibitors, and ICIs, stems from analyses of variant histology trials. The ASPEN trial, which compared sunitinib versus everolimus in nccRCC patients, included 16 chRCC patients demonstrating a more favorable response profile to everolimus versus sunitinib. The mPFS for chRCC patients treated with sunitinib was 5.5 months (80% CI 3.2–19.7) versus 11.4 (80% CI 5.7–19.4) months in those treated with everolimus (HR 0.7 (80% CI 0.3–1.7)). 35 It should be noted that the mutational burden within chRCC is low, potentially hinting at a mechanistic rationale for the favorable response to mTOR inhibition. Additionally, Voss et al., 32 in a phase II trial evaluating bevacizumab plus everolimus, observed prolonged disease control in chRCC patients, with three out of five patients remaining on treatment for over 12 months. Furthermore, the combination of anti-VEGF TKIs plus mTOR inhibitors has shown increased antitumor activity in chRCC compared to pRCC. For example, Hutson et al. 61 demonstrated in their single-arm, phase II trial with lenvatinib plus everolimus that the ORR and mPFS for chRCC were 44% and 13.1 months versus 15% and 9.1 months in pRCC patients.
While the introduction of immunotherapy has revolutionized the RCC treatment landscape, chRCC is one histological subtype with scarce responses to ICIs.42,45,62 Due to a low proportion of immune infiltrating cells and minimal expression of antigen-presenting genes, it is thought chRCC has an “immunologically cold” tumor microenvironment. 63 In a phase II trial conducted by Lee et al. 46 evaluating nivolumab plus cabozantinib, no responses were observed among the seven chRCC patients. COSMIC 021, a phase II trial evaluating the combination of atezolizumab and cabozantinib, reported one response out of nine chRCC patients. Similarly, KEYNOTE-427, a phase II trial evaluating monotherapy pembrolizumab in nccRCC, demonstrated an unfavorable ORR of 9.5% in chRCC patients. 42 More recently, KEYNOTE-B61 reported data on lenvatinib plus pembrolizumab in nccRCC, of which 29 chRCC patients were included with an ORR of 28%. 6 Although the response rate for chRCC was considerably lower than other histological subtypes, there was a notable improvement in ORR versus KEYNOTE-427.
Collecting duct carcinoma
Collecting duct carcinoma (CDC) is a rare, aggressive carcinoma that arises from the renal medullary collecting ducts. Like chRCC, CDCs are not graded based on histopathologic criteria; by definition, they are high-grade tumors. 64 They typically present at an advanced stage, with a significant portion of cases already showing local invasion or distant metastasis at the time of diagnosis. In a report of 81 patients, 57% of the patients presented with pT3 or pT4 disease, and 32% had distant metastasis at presentation. 65
Given the rare nature of CDC, the molecular landscape of this aggressive carcinoma has not been fully understood. A study by Pal et al. explored the genomic profiling of 17 CDC patients and identified several recurrent GAs. The most common GAs were in NF2 (29%), SETD2 (24%), SMARCB1 (18%), and CDKN2A (12%). This study highlighted the potential therapeutic implications of these findings, suggesting that mTOR inhibitors might benefit patients with NF2 alterations. Additionally, the study noted that alterations in FH and SMARCB1 occurred in a mutually exclusive manner to NF2 alterations, indicating distinct molecular pathways in CDC pathogenesis. 66
Another study by Wang et al. performed whole-exome sequencing (WES) and transcriptome sequencing on seven CDC samples and identified a CDKN2A homozygous deletion in three samples. This study also confirmed the frequent loss (62.5%) of CDKN2A expression through FISH screening of additional samples. The study found that CDKN2A loss was associated with the upregulation of multiple solute carrier (SLC) family genes, which have a role in drug resistance. 67 Notably, SLC7A11, a cystine transporter associated with cisplatin resistance, was overexpressed in 80% of CDC cases. 68 This suggests that targeting SLC7A11 could be a potential therapeutic strategy for overcoming cisplatin resistance in CDC patients. 67
Historically, CDC and urothelial carcinoma were believed to have similar clinical and pathological characteristics, partly due to shared anatomical proximity between the renal collecting ducts and the urothelial tract within the renal system. 69 This led clinicians to adopt treatment regimens initially developed for urothelial carcinoma in CDC management. Orsola et al. 70 previously highlighted overlapping clinical features between these malignancies, supporting this initial rationale. However, more recent molecular studies, including comprehensive genomic analyses by Pal et al. 66 and transcriptomic profiling by Malouf et al., 71 have demonstrated substantial genetic and transcriptomic differences between CDC and urothelial carcinoma, underscoring their distinct biological nature and unique molecular drivers. These findings suggest that future therapeutic strategies should be tailored specifically to the distinct genetic and molecular traits of each cancer type.
Management
Due to similar clinical features between CDC and urothelial carcinoma, several phase II trials have examined the use of platinum-based cytotoxic chemotherapy in CDC. Oudard et al. were the first to evaluate gemcitabine plus cisplatin or carboplatin in patients with previously untreated CDC. The trial included 23 patients with an ORR of 26%, mPFS of 7.1 months, and median overall survival (mOS) of 10.5 months. 72 Given relatively limited responses with chemotherapy in CDC and the introduction of multikinase inhibitors in ccRCC, Sheng et al. investigated the combination of sorafenib plus gemcitabine and cisplatin in CDC. The results were slightly more favorable, with an ORR of 31%, mPFS of 8.8 months, and mOS of 12.5 months. 73 The BONSAI trial, a phase II study evaluating first-line cabozantinib in CDC, demonstrated that CDC can be sensitive to VEGF inhibitor monotherapy with a reported ORR of 35%. 74 Based on promising data from urothelial carcinoma, the addition of bevacizumab, a VEGF-A monoclonal antibody, to platinum-based chemotherapy was hypothesized to improve outcomes in CDC. The BEVABEL-GETUG phase II trial incorporated 34 patients with either CDC or RMC (31 CDC patients), reporting an ORR of 41%, mPFS of 5.9 months, and mOS of 11.1 months. 75 Of note, the trial enrollment was ended prematurely due to limited efficacy and toxicity with two treatment-related fatalities.
Currently, there is a paucity of clinical data supporting the use of ICIs in CDC. Key immunotherapy trials in nccRCC either reported small sample sizes or excluded CDC due to its aggressive phenotype, typically requiring platinum-based chemotherapy. Several case reports have reported the safety and efficacy of nivolumab in CDC. To date, there are no ongoing studies examining the use of immunotherapy in CDC. Based on preliminary data from the CICERONE trial (NCT05372302) showing 30% nectin-4 expression in CDC, the REPRINT trial (NCT06302569), a single-arm, phase II trial evaluating pembrolizumab plus enfortumab vedotin in CDC, has been designed and is currently awaiting recruitment.
Molecularly defined RCCs
MiT family translocation RCC
Translocation renal cell carcinomas (tRCC) are rare tumors that typically present at a younger age and exhibit an aggressive course compared to ccRCC. In a study that characterizes adolescent and pediatric RCC, 46% of the pediatric RCCs were translocation associated. 76 In the 2016 WHO classification of renal tumors, TFE3 and TFEB rearranged RCCs were grouped under the MiT family of RCC (MiTF-RCC). However, in the 2022 classification, these two entities were recognized as distinct subtypes of RCC. 2
The MiT family includes MiTF, TFEB, TFE3, and TFEC transcription factors. The TFE3 gene, located on chromosome Xp11.2, can undergo translocation, fusing with one of over 20 identified genes. This induces the expression of a proto-oncogene coding for an RTK, triggering downstream signaling pathways that promote uncontrolled cell proliferation. 77 The specific gene that fuses with TFE3 also impacts the tumor’s phenotype and influences patient outcomes. Tumors with the TFE3-ASPSCR1 fusion were identified to be highly aggressive, defined by a higher nuclear grade, more frequent metastasis, and shorter overall survival. 78
In addition to these translocations, multiple other genomic alterations have been identified in tRCC. WES has shown that tRCCs tend to have a lower tumor mutational burden compared to ccRCC and pRCC. 78 However, chromosome copy number alterations were found to be similar to ccRCC or pRCC. The most common chromosome copy alteration was a gain of 17q (44%), followed by gains of chromosomes 12 (37%) and 7 (31%), losses of chromosomes 9p (37%), 3p, 1p, 17p, 18q (each observed in 31% of the patients). 79
The need to develop molecular-based targeted therapies led Baba et al. to employ a mouse model. Their study revealed high tyrosine kinase receptor RET expression levels in TFE3 translocated models. They also studied vandetanib, an RET inhibitor, and demonstrated statistically significant reduced tumor growth in the vandetanib-treated group compared to the control group. 80 To identify potential targets, Lang et al. employed a high-throughput drug screen of TFE3-RCC cell lines. They found several potential pharmacological agents, including PI3K/mTOR pathway inhibitors, histone deacetylases, and Src/Abl kinases. Notably, the PI3K/mTOR inhibitor NVP-BGT226, the transcription inhibitor Mithramycin A, and the GPNMB-targeted antibody–drug conjugate CDX-011 demonstrated significant efficacy in preclinical models. 81 Interestingly, multiple studies have previously identified GPNMB as a diagnostic marker.80,82 One of the studies also underscored the potential of GPNMB as a therapeutic target. 82
In addition to these advancements, several other biomarkers and potential therapeutic targets became apparent. TRIM63 has emerged as a sensitive and specific biomarker for MiTF-RCC, showing significant overexpression in TFE3 and TFEB translocation RCCs compared to other renal tumor subtypes. 83 TFE3-splicing factor (TFE3-SF) fusions have also been identified as functional drivers in MiTF-RCC. 84 These findings collectively underscore new diagnostic markers and therapeutic strategies that could improve the management of these aggressive cancers.
Multiple studies investigated the immune landscape and checkpoint inhibition efficacy in tRCC. 85 Sun et al.’s study suggests that the fusion subtype may play a role in the tumor immune microenvironment (TIME). MED15-TFE3 fusion exhibited much higher expression of PD-L1 (~80%, n = 8) compared to other fusion types (~20%, n = 55). They also reported that tRCC has a lower immune infiltration score and T-cell infiltration score when compared to TCGA’s ccRCC cohort. In their report, 63% of the patients exhibited type 1 TIME, characterized by a lack of tumor-infiltrating lymphocytes and PD-L1 expression.78,86 In contrast, Bakouny et al. 87 reported that tRCC contains an appreciable density of tumor-infiltrating CD8+ T cells and points to notable responses to immune checkpoint inhibition. These findings suggest that the fusion type could be a factor in guiding future trials involving ICIs for tRCC.
Management
As with other nccRCC histological subtypes, no dedicated clinical trials exist for tRCC, making available data from retrospective studies the primary driver for clinical management decisions. Initially, a few retrospective studies helped establish the safety and efficacy of sunitinib, a targeted VEGF inhibitor, in tRCC with a reported ORR of 10%–15% and mPFS of 7–8 months.88,89 Of note, a limitation of these earlier studies was the use of immunohistochemistry, possibly incorporating study participants without a genomic translocation. More recently, elucidating MET expression in tRCC sparked interest in evaluating the use of cabozantinib within tRCC. Thouvenin et al. 90 conducted an international, multicenter, retrospective cohort study including 52 patients (TFE3: 46 patients and TFEB: 6 patients) with previously treated or untreated metastatic tRCC treated with cabozantinib monotherapy. Among evaluable patients, the ORR was 17.3% (two complete responses and seven partial responses), mPFS was 6.8 months, and mOS was 18.3 months. Given the unique characteristics of tRCC tumor microenvironment, there is growing interest in understanding the role of ICIs in its management. One retrospective study evaluated tRCC patients who received ICI monotherapy. Of the 24 patients included, the ORR was 16.7%, and mPFS was 2.5 months. 85 In parallel with the current standard of care for ccRCC, combination therapy (dual ICI or ICI + VEGF inhibitors) was subsequently evaluated in one retrospective study with 29 patients. This study reported more favorable outcomes in patients treated with ICI + VEGF versus dual ICI inhibitors with an ORR 36% versus 5.5%, mPFS 5.4 versus 2.8 months, and mOS 30.7 versus 17.8 months, respectively. 91 KEYNOTE-B61, the largest combination therapy (pembrolizumab plus lenvatinib) dataset in previously untreated nccRCC, enrolled six patients with tRCC reporting an ORR of 67% (four partial responses). 6 The development of NCT03595124 sparks the advent of the first prospective tRCC trial: randomizing patients to axitinib plus nivolumab, axitinib alone, or nivolumab alone.
SMARCB1-deficient medullary RCC
SMARCB1-deficient medullary RCC, or historically RMC, is a rare, distinct tumor almost always associated with sickle cell trait disease. 92 SMARCB1 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1) is one of more than 15 subunits of the SWI/SNF chromatin remodeling complex. 28 SMARCB1 loss has been identified in several cancers, such as malignant rhabdoid tumors and atypical teratoid rhabdoid tumors, collectively referred to as SMARCB1-deficient cancers. While we still do not fully understand SMARCB1 and its functions, recent studies have pointed out that SMARCB1 plays a role in regulating oncogenic enhancers and promoters. When SMARCB1 is lost, these regulatory regions become inactive, leading to genome-wide transcriptional dysregulation.93,94
SMARCB1-deficient medullary RCC primarily affects young Black/African American adults with sickle cell trait disease, with a mean age of diagnosis reported as 28 in a study utilizing the SEER database. 95 Its clinical features are highly aggressive. Patients usually present with high-stage tumors and have metastatic disease. A multicenter collaborative study investigated the management and survival outcomes of RMC, reporting a 12-month overall survival rate of 53% (95% CI: 40–69) and a 24-month overall survival rate of 17% (95% CI: 9–33). 96
Under normal conditions, the renal medulla is the most hypoxic and hypertonic tissue in the human body, which is necessary for urine concentration. However, this environment can lead to sickling in individuals with sickle cell trait, further exacerbating the hypoxic conditions in the tissue. Red blood cell sickling-induced ischemia in the renal medulla has been hypothesized to be crucial in the pathogenesis of SMARCB1-deficient medullary RCC. 92 Soeung et al. suggested that hypoxia causes a decrease in SMARCB1 protein levels in renal cells as a natural response, protecting the cells from hypoxic stress. While the reduction of SMARCB1 protein levels protects renal cells from hypoxia, sickling prolongs and intensifies the hypoxia in the renal medulla. Prolonged hypoxia ultimately allows somatic mutations to occur and expand, resulting in a complete loss of the tumor suppressor SMARCB1, thus driving tumorigenesis. 97
Management
Currently, there is no standard treatment for managing RMC. Due to RMC’s aggressive phenotype and limited responses to standard VEGF inhibitors, the traditional approach incorporates cytotoxic chemotherapy regimens such as MVAC (methotrexate, vinblastine, doxorubicin, and cisplatin) or PCG (paclitaxel, cisplatin or carboplatin, and gemcitabine). However, no proposed chemotherapy regimen has shown superiority, highlighting the need for innovative therapeutic strategies. Preclinical studies have shown that RMC is contingent upon SMARCB1 inactivation, which can upregulate protein anabolism and render cells vulnerable to proteasome inhibition. 98 Several case reports documented the use of bortezomib, a first-generation proteasome inhibitor, in RMC as monotherapy or in combination with cytotoxic chemotherapy agents, noting more promising results in combination therapy.99,100 This led to the development of NCT03587662, a phase II study investigating proteasome inhibition in RMC with ixazomib, a second-generation proteasome inhibitor, plus gemcitabine and doxorubicin. The trial is currently active, with plans for completion in 2025. Prior studies have noted EGFR upregulation in RMC, prompting Wiele et al. to report a retrospective experience with erlotinib, an EGFR inhibitor, plus bevacizumab, a VEGF inhibitor, in 10 RMC patients. Two patients achieved partial responses, and seven had stable disease, but of note, patients were heavily pretreated with a median of 2.5 prior systemic therapies. 101 Msaouel et al. 102 previously reported the comprehensive molecular and immune profiling of RMC, highlighting a heterogeneous tumor microenvironment of PD-L1 expression and tumor-infiltrating lymphocytes. Case reports have shown conflicting experiences with PD-1 inhibition using nivolumab in RMC, 103 prompting the recent reporting of the first prospective evaluation of single-agent pembrolizumab in RMC. Six patients with advanced disease were enrolled with no objective responses observed and rapid disease progression within 13 weeks. 104 Although this trial was nonrandomized and had limited numbers, it suggests that PD-1 inhibitor monotherapy has limited clinical activity in RMC. These studies emphasize the need for additional targeted and immunomodulatory strategies to provide antitumor activity in RMC. Currently, there are three ongoing clinical trials examining innovative strategies in RMC: (1) NCT03274258—efficacy of nivolumab in combination with the CTLA-4 inhibitor ipilimumab specifically in patients with RMC, (2) NCT05347212—efficacy of nivolumab in combination with high doses of the LAG-3 inhibitor relatlimab, and (3) NCT03866382—efficacy of cabozantinib in combination with nivolumab and ipilimumab in rare genitourinary malignancies including RMC.
Fumarate hydratase-deficient RCC
fhRCC was first identified in individuals with hereditary leiomyomatosis and renal cell cancer (HLRCC). HLRCC is linked to germline mutations in the fumarate hydratase (FH) gene and is characterized by benign uterine and cutaneous leiomyomas alongside aggressive RCCs. In the 2022 WHO classification of renal cancers, HLRCC was redefined as fhRCC, acknowledging its association with both germline and somatic mutations in the FH gene. 2
FH is an enzyme in the tricarboxylic acid cycle, crucial for generating ATP through mitochondrial oxidative phosphorylation. Mutations in the FH gene lead to the accumulation of fumarate, disrupting energy production and impairing the activity of histones and DNA demethylases, which results in aberrant gene expression. 105
Multiple studies have explored the immunogenicity of FH-deficient RCC. Sun et al. reported that half of the evaluated samples exhibited type 2 TIME, characterized by positive PD-L1 expression and CD8+ T-cell infiltration. They suggested that antitumor immunity in FH-deficient RCC might be blocked by a PD-1/PD-L1-mediated immune escape mechanism. 106 Therefore, blocking PD-1/PD-L1 could be beneficial in treating FH-deficient RCCs. A study exploring genomic features between primary and metastatic sites found that primary tumors and metastatic lesions were highly immunogenic. Results showed high PD-L1 expression on primary (84%) and metastatic lesions (93%). While both sites show high PD-L1 expression, metastatic sites had higher T cells, co-activation molecules, antitumor cytokine signatures, and numerically higher Th1 cells. 107 Chen et al. recently proposed an immune signature to select optimal candidates for ICI and TKI combination treatment. They also compared single-agent TKI therapy with ICI and TKI combination therapy and found the latter more favorable. However, their cohort had no patients treated with erlotinib and bevacizumab combination. 108
To shed light on the genomic background of FH-deficient RCC, Xu et al. identified 77 patients with FH-deficient RCC; 70 had germline deficiency, and 7 had somatic deficiency. Their study focused on genomic profiling, clinical characterization, and treatment efficacy, assessing treatment response to either a combination of bevacizumab and erlotinib (12/67, 18%), TKI monotherapy (29/67, 43%), or a combination of ICI and TKI (26/67, 39%). The combination of ICI and TKIs was found to be associated with a more favorable OS (HR 0.19; 95% CI 0.04–0.90, p = 0.019) and PFS (HR 0.22; 95% CI 0.07–0.71, p = 0.005) when compared to the bevacizumab and erlotinib combination. 109 Notably, they observed 25% ORR with first-line bevacizumab and erlotinib combination, while the phase II trial of the combination showed a promising 64% ORR. 110
Exploring new therapeutic targets in FH-deficient RCC, a recent study by Noronha et al. devised a promising therapeutic strategy by targeting the silencing of the NAPRT protein expression in FH-deficient RCC. In FH-deficient tumors, the accumulation of fumarate leads to global DNA hypermethylation, including hypermethylation of CpG islands in the NAPRT promoter, resulting in its silencing. 111 This disruption impairs the NAD+ biosynthesis pathway, forcing cancer cells to rely heavily on the salvage pathway for NAD+ production. Consequently, these cells become highly susceptible to nicotinamide phosphoribosyl transferase inhibitors (NAMPTis), which target the salvage pathway. 112 A simplified visualization of NAD+ production pathways is presented in Figure 3.

Simplified visualization of NAD+ production pathways. Mutations in the FH gene result in the accumulation of fumarate, leading to global DNA hypermethylation and subsequent silencing of NAPRT. FH-mutant tumor cells rely on the NAD+ salvage pathway for NAD+ production. Inhibiting NAMPT disrupts this pathway and induces tumor cell death. Additionally, PARP inhibitors can be used in combination with NAMPT inhibitors for synergistic tumor cell killing. The five arrows in the de novo synthesis pathway indicate the five additional steps to synthesize quinolinic acid from tryptophan.
In preclinical models, NAMPT inhibitors effectively depleted NAD+ levels in NAPRT-deficient cells, leading to metabolic stress and cell death. Furthermore, combining NAMPT inhibitors with PARP inhibitors (PARPi) resulted in synergistic tumor cell death by disrupting both NAD+ metabolism and DNA repair mechanisms. This therapeutic approach not only identifies NAPRT as a key biomarker in FH-deficient RCC but also reveals a new potential for combination therapies that target both metabolic vulnerabilities and DNA repair mechanisms. As a result, NAMPT inhibitors, alone or in combination with PARPi, represent a promising targeted treatment strategy for patients with FH-deficient RCC. 112
NAD metabolism was also investigated by Najera et al. in FH-deficient HLRCC cell lines using OT-82, a novel NAMPT inhibitor. Their study revealed significantly elevated expression of NAD+ salvage pathway enzymes in both in vitro and patient-derived HLRCC cell lines, with NAMPT expression being at least four times higher in HLRCC tumor lines compared to normal kidney tissue (p < 0.001). NAMPT inhibition with OT-82 depleted NAD metabolites and reduced protein PARylation, resulting in growth suppression in both cell lines and xenografts. The authors also investigated the rescue with NMN, a precursor substrate for NAD production. In their study, cell lines treated with OT-82 demonstrated significant loss of proliferation and decreased viability. However, co-treatment with NMN effectively restored cell proliferation and rescued both PAR and NADP+/NADPH levels, confirming that OT-82 specifically targets NAD+-dependent processes. 113
Although NAMPT targeting is not a new concept in oncology, previous trials of NAMPT inhibitors in sporadic tumors yielded limited efficacy.114,115 However, the studies by Noronha et al. and Najera et al. have demonstrated that NAMPT inhibition shows higher specificity and efficacy in FH-deficient tumors, where the metabolic vulnerability due to disrupted NAD+ biosynthesis creates a unique therapeutic opportunity.
Management
Treatment for fhRCC is often complex due to its limited responsiveness to traditional RCC therapies. 116 Studies have indicated promising responses from combining antiangiogenic agents with immunotherapy, notably bevacizumab paired with erlotinib, which has shown beneficial response rates in specific cohorts.110,117 Ongoing clinical trials are exploring combinations of immune inhibitors and targeted therapies to further improve outcomes in advanced FH-deficient RCC. Additionally, a recent phase II trial involving the combination of talazoparib, a PARPi, and avelumab, an immune checkpoint inhibitor, indicated that this regimen is well tolerated, although efficacy results in terms of response rates were limited. 118 Further clinical trials are underway to assess the effectiveness of pairing ICIs with targeted therapies, aiming to improve survival outcomes in patients with advanced FH-deficient RCC. Given the malignancy’s aggressive nature, patients who have undergone surgical resection are advised to undergo intensive follow-up imaging every 3 months for the first 2 years, ensuring early detection of potential recurrence or distant metastasis. 116
Unclassified RCC
Unclassified renal cell carcinoma (uRCC) refers to renal tumors whose histology and morphology do not fit into any recognized subtype. This broad category includes tumors with mixed features of known subtypes, unclassified oncocytic neoplasms, or tumors that present purely sarcomatoid histology. Managing uRCC poses significant diagnostic and therapeutic challenges, as it is a diagnosis of exclusion and lacks established standard treatments.
To address the uncertainties surrounding uRCC, several studies have investigated the molecular and genetic characteristics of these tumors. In a cohort of 62 patients, Chen et al. 119 identified mutations in NF2 (18%), SETD2 (18%), and BAP1 (13%). Interestingly, the rate of NF2 mutations was notably higher in this cohort when compared to other renal carcinoma subtypes studied in the TCGA database. While VHL mutations are found in roughly 75% of ccRCC cases, only one patient in their study exhibited this mutation. A separate analysis using data from the American Association for Cancer Research’s Genomics Evidence Neoplasia Information Exchange database revealed VHL mutations in 12% of patients (22 out of 184). Furthermore, NF2 and SETD2 mutations were observed in 16% of patients, aligning with prior studies. Notably, NF2 alterations were linked to worse mOS in this study, 30.7 compared to 87.1 months (p = 0.058). 120 Although both studies had relatively small patient groups, the consistent presence of NF2 mutations is notable across the datasets, suggesting that NF2 loss may define a distinct molecular subset of RCC and offer potential novel therapy options.
Management
Therapeutically, prospective studies specifically targeting uRCC are sparse. However, data from basket trials suggest some efficacy with TKIs and ICIs. For instance, pembrolizumab demonstrated a 30.8% ORR in a subset of uRCC patients in the KEYNOTE-427 trial, with an mPFS of 2.8 months and mOS of 17.6 months. 42 Similarly, sunitinib and cabozantinib have shown modest response rates in retrospective studies, with mPFS ranging from 3.2 to 6 months.121,122 These findings underscore the need for further exploration of molecular drivers and targeted therapies to optimize treatment strategies for this challenging subtype.
Discussion
The reclassification of nccRCC by the WHO in 2022 represents a significant advancement in the field. This new classification emphasizes molecular and genetic markers, allowing for a more specialized approach to diagnosing and treating nccRCC. The shift toward a molecular classification system facilitates the development of targeted therapies and personalized treatment plans, which are essential for improving patient outcomes. Additionally, this molecular approach enables more precise patient stratification in clinical trials, potentially leading to therapeutic strategies that are both more effective and tailored to the unique biology of each subtype.
Despite these advancements, several challenges remain in the research and treatment of nccRCC. The relative rarity and molecular heterogeneity of nccRCC limit the feasibility of conducting large-scale randomized clinical trials, which are the gold standard for evaluating treatment efficacy. As a result, current treatment regimens are often adopted from those for ccRCC, but they do not optimally address the distinct genetic and molecular drivers characteristic of nccRCC. This underscores the need for developing targeted therapies that specifically address the diverse molecular and genetic alterations found within nccRCC subtypes. An overview of ongoing and completed clinical trials in nccRCC is presented in two tables: Table 1 summarizes basket trials that included patients with nccRCC, while Table 2 highlights histology-specific studies.
Clinical trials enrolling multiple non-clear cell renal cell carcinoma subtypes.

ICI, immune checkpoint inhibitor; CR, complete response; mAb, monoclonal antibody; mPFS, median progression-free survival; nccRCC, non-clear cell renal cell carcinoma; ORR, objective response rate; OS, overall survival; PR, partial response; TKI, tyrosine kinase inhibitor.
Clinical trials enrolling histology-specific non-clear cell RCC subtypes.

ADC, antibody-drug conjugate; CDC, collecting duct carcinoma; Chemo, chemotherapy; CR, complete response; FH-deficient RCC, FH-deficient renal cell carcinoma; ICI, immune checkpoint inhibitor; mAb, monoclonal antibody; mPFS, median progression-free survival; nccRCC, non-clear cell renal cell carcinoma; ORR, objective response rate; OS, overall survival; PR, partial response; pRCC, papillary renal cell carcinoma; RMC, renal medullary carcinoma; TKI, tyrosine kinase inhibitor; Translocation, translocation-associated renal cell carcinoma; VEGFR2, vascular endothelial growth factor receptor 2.
Future research should focus on further explaining the genetic and molecular landscape of nccRCC. Comprehensive genomic studies are needed to identify additional therapeutic targets and develop novel treatment strategies. Collaborative efforts between researchers and clinicians are essential to advancing the understanding and treatment of nccRCC. Expanding access to NGS and other molecular profiling tools may facilitate broader identification of actionable mutations, allowing for more widespread use of precision medicine approaches. Another critical area of need is improving access to clinical trials. Currently, clinical trials are often limited to patients who live near cancer centers or those with sufficient financial resources, leaving many patients underserved. Addressing these barriers by expanding the geographic reach and accessibility of trials is essential to ensuring that all patients, regardless of location or financial situation, can benefit from the latest advancements in nccRCC treatment.
In summary, the recent advancements in the classification and understanding of nccRCC have significant implications for clinical practice and research. The shift toward a molecular classification system and the development of targeted therapies represent important steps forward. However, ongoing research and collaborations are necessary to fully realize the potential of these advancements and improve patient outcomes.