
Abstract
Background:
Neuroblastoma (NB) is rare in adolescents and adults, resulting in limited availability of data.
Objectives:
We comprehensively investigated the characteristics, treatments, and outcomes of adolescent and adult patients with NB, aiming to provide a more in-depth insight into this disease.
Design:
A retrospective, single-center study.
Methods:
We retrieved and analyzed the medical data of patients with NB aged 10 years or older at diagnosis who were treated at Sun Yat-sen University Cancer Center between June 2005 and January 2024.
Results:
Sixty-five patients (30 males and 35 females) were enrolled, with a median age of 20 years (interquartile range, 14–26 years), including 27 patients aged 10–18 years and 38 patients aged >18 years. Most patients were classified as M-stage disease (n = 40, 61.5%), high-risk (n = 42, 64.6%), and poorly differentiated NB (n = 27, 41.5%). Additionally, 3 (6.7%) patients had MYCN amplification, and 5 (25%) had ALK mutations. The genomic landscape revealed that mutations in the cell cycle and DNA repair pathways are related to chemotherapy sensitivity. After induction therapy, 34 (52.3%) patients achieved complete response (CR). The 5-year progression-free survival (PFS) and overall survival (OS) rates were 33.1% ± 6.9% and 55.1% ± 7.6%, respectively. Patients who achieved CR after induction therapy had superior PFS (p = 0.009), with 5-year PFS rates of 44.0% ± 10.6% compared to 18.5% ± 8.5% in non-CR patients.
Conclusion:
Adolescent and adult patients with NB exhibit distinct characteristics, less chemotherapy sensitivity, and poorer outcomes compared to pediatric patients. Achieving CR after induction therapy is associated with better outcomes. Further investigation for new therapies is required.
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in early childhood, constituting approximately 10% of all malignant tumors in children. 1 Typically, the median age at diagnosis is 18 months, with more than 95% of tumors occurring before 10 years of age. 2 Age at diagnosis is a prognostic factor for NB. 3 Older age is a prognostic factor of poor survival, with outcomes gradually worsening with increasing age at diagnosis. 4 Newborn patients aged <18 months generally have better outcomes, with some experiencing spontaneous regression of tumors, whereas adolescents and adults tend to have an unfavorable prognosis. 5
Adolescents and adults rarely develop NB, resulting in limited clinical and biological data in the literature. A limited series of reports have revealed distinct characteristics and biological features of NB in adolescent and adult populations: more indolent behavior, a longer disease course, predominance of advanced stages, unusual pattern of metastatic localizations, lower urine catecholamine levels, higher incidence of unfavorable histology, rare MYCN amplification, less sensitivity or poor tolerance to pediatric chemotherapy regimens, and generally worse outcomes.6–17 The genetic landscape of adolescent and adult NB patients remains poorly characterized. Limited results from targeted exome sequencing found a high frequency of loss-of-function mutations in ATRX and ALK, as well as less common mutations in genes such as BRCA1, MUTYH, ERBB3, MAGI2, RUNX1, MLL.17,18 Currently, standard NB treatments are not specifically adapted to the unique pathophysiology of adolescent and adult patients. Given the poor outcomes and limited understanding of optimal treatment strategies for this rare population, comprehensive clinical and biological data are urgently needed to guide future research and improve therapeutic approaches.
Herein, we retrospectively retrieved and analyzed the medical data of patients with NB aged ≥10 years at the time of diagnosis to provide a more in-depth insight into the clinical characteristics, treatment modalities, and prognostic data of this rare disease in adolescents and adults.
Materials and methods
Patients
We reviewed all medical records of patients with NB aged ⩾10 years treated at Sun Yat-sen University Cancer Center (SYSUCC) between June 2005 and January 2024.
This study was approved by the Institutional Review Board and Ethics Committee of the SYSUCC (B2024-504-01) and was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans and Good Clinical Practice. The requirement for written informed consent was waived owing to the study’s retrospective nature. The reporting of this study conforms to the STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) statement 19 (Supplemental File 1).
Diagnosis and risk stratification
The diagnosis of NB was confirmed by histopathology or the demonstration of bone marrow involvement with extrinsic cells, accompanied by elevated urinary catecholamine metabolites. Workup at the initial diagnosis and subsequent response assessment were as follows: urinary homovanillic acid, vanillylmandelic acid, bilateral bone marrow aspiration and core biopsy, computed tomography (CT), magnetic resonance imaging, I123 metaiodobenzylguanidine (MIBG) scans, and fluorodeoxyglucose-positron emission tomography scans (PET/CT, if non-MIBG avid). The MYCN status was detected by fluorescence in situ hybridization (FISH) or by next-generation sequencing (NGS). Status by FISH is defined as follows: not amplified (2 copies per cell), gain (>2–8 copies per cell or <4-fold increase), amplified (>8 copies per cell or ⩾4-fold increase, can be >30-fold increase). The staging system and risk stratification were based on the International Neuroblastoma Risk Group (INRG) Staging System 20 and the INRG classification system. 21
Genomic profiling and circulating tumor DNA surveillance
We performed comprehensive genomic profiling using whole exome sequencing (WES) on paired samples from 12 patients. Genomic DNA was extracted from the formalin-fixed, paraffin-embedded tumor samples and matched white blood cells. After DNA extraction, 1 ng DNA was subjected to polymerase chain reaction to amplify Housekeeping gene fragments using a set of primers (synthesized by Invitrogen, Life Technologies, Carlsbad, CA, USA). The amplified products were then fragmented into approximately 250 base pairs using sonication. Sample library preparation was performed using VAHTSTM Universal DNA Library Prep Kit for Illumina® V3 (Vazyme Biotech, Nanjing, China). Target enrichment was done by NGS-based hybrid capture method using the pool of customized individually synthesized plus xGen Exome Research Panel. Hybridization capture using target gene panel and xGen® Lockdown® Reagents followed the manufacturer’s protocol, in order to enrich for the targeted nearly 20,000 genes. Post-capture libraries were pooled together, denatured, and diluted to 200–250 pM for sequencing on Illumina NovaSeq 6000 sequencing platform (Illumina, San Diego, CA, USA). Various genomic alterations, including single nucleotide variants (SNVs), insertions and deletions, copy number variations, and gene rearrangements and fusions, were subsequently analyzed.
Longitudinal circulating tumor DNA (ctDNA) surveillance was conducted in six patients at multiple time points, including baseline, every 2 months during treatment (if available), at the end of treatment, and every 6 months post-treatment. Briefly, peripheral blood samples were collected and processed for cell-free DNA extraction, followed by DNA library preparation and high-throughput sequencing. Molecular tracking and variant calling for SNVs were subsequently performed. Samples with three or more detected variants were classified as ctDNA-positive.
Treatment protocol and response assessment
Treatment followed pediatric protocols, with dosage adjustments based on patient tolerance. For the very low-risk- and low-risk groups, the primary treatment was surgery and chemotherapy using the CAV (cyclophosphamide, pirarubicin, vincristine)/EP (etoposide, cisplatin) regimen. Intermediate-risk patients underwent surgery, chemotherapy (CAV/EP regimen), and radiotherapy. For high-risk patients, the treatment regimen included surgery, chemotherapy using the CAV (cyclophosphamide, pirarubicin, vincristine)/VIP (etoposide, ifosfamide, cisplatin) regimen, ±autologous stem cell transplantation (ASCT), or radiotherapy, followed by maintenance therapy, which included anti-GD2 immunotherapy, cis-retinoic acid, and 1-year metronomic chemotherapy (cyclophosphamide, vinorelbine, etoposide, celecoxib).
The overall response was assessed using the International Neuroblastoma Response Criteria: complete response (CR), partial response (PR), mixed response (MR), stable disease (SD), and progressive disease (PD). 22
Statistical analysis
All statistical analyses were performed with SPSS version 25 (IBM Corp., Armonk, NY, USA). Progression-free survival (PFS; time to disease relapse or progression) and overall survival (OS; time to death from any cause or the last follow-up) were assessed from the time of diagnosis. Survival analysis was performed using the Kaplan–Meier method and compared using the log-rank test. A Cox regression model was used to assess potential factors that could predispose patients to survival. Pearson’s Chi-square test was used to compare clinical and biological characteristics between the adolescent and adult groups. p < 0.05 indicated statistical significance.
Results
Characteristics of the patients and clinical features at diagnosis
A total of 65 patients (30 males and 35 females), aged 10–68 years old at the time of diagnosis, were analyzed. The median age at diagnosis was 20 years (interquartile range (IQR), 14–26 years), with 27 patients aged 10–18 years and 38 patients aged >18 years. The baseline characteristics and clinical features are summarized in Table 1, and the detailed clinical information for each patient is listed in Supplemental Table S1.
Summary of patient characteristics.
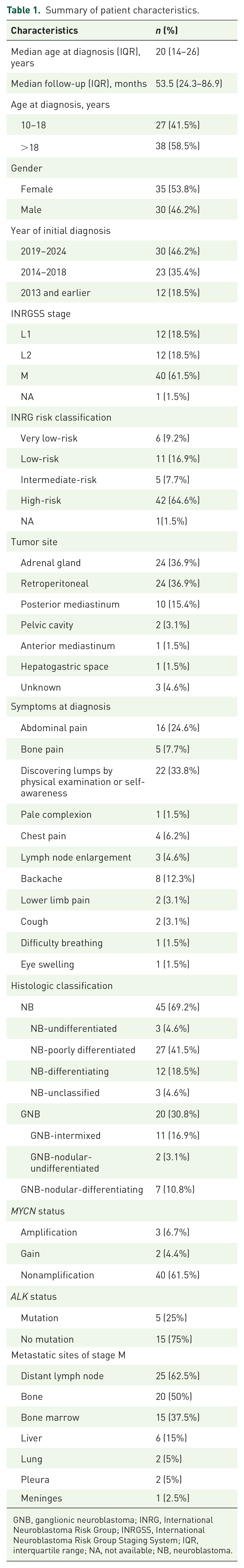
GNB, ganglionic neuroblastoma; INRG, International Neuroblastoma Risk Group; INRGSS, International Neuroblastoma Risk Group Staging System; IQR, interquartile range; NA, not available; NB, neuroblastoma.
The primary tumor sites were the adrenal gland (n = 24, 36.9%), retroperitoneum (n = 24, 36.9%), and mediastinum (n = 10, 15.4%). At initial diagnosis, 40 (61.5%) patients exhibited distant tumor metastasis, with the most common sites of metastasis being the lymph nodes (n = 25, 62.5%), bone (n = 20, 50%), and bone marrow (n = 15, 37.5%). Among the 65 patients, 22 (33.8%) were initially diagnosed with NB based on the discovery of a mass during routine physical examination or self-awareness. The sites of the primary tumor and metastases correlated with the most common presenting symptoms at diagnosis, including abdominal pain (n = 16, 24.6%) and backache (n = 8, 12.3%). Twelve (18.5%) patients presented with L1-stage, 12 (18.5%) patients with L2-stage, and 40 (61.5%) patients with M-stage of the disease. Most patients were classified into the high-risk group (n = 42, 64.6%), whereas others were classified into the very low-risk (n = 6, 9.2%), low-risk (n = 11, 16.9%), and intermediate-risk (n = 5, 7.7%) groups.
Biological features and genetic landscape
Regarding histological category, 45 (69.2%) patients were diagnosed with NB (including 12 differentiating NB, 27 poorly differentiating NB, 3 undifferentiated NB, and 3 unclassified), and 20 (30.8%) patients were diagnosed with ganglionic neuroblastoma (GNB, including 11 intermixed GNB, 7 nodular-differentiating GNB, and 2 nodular-undifferentiated GNB).
MYCN oncogene evaluation results were available for 45 patients, revealing 3 (6.7%) patients with MYCN gene amplification and 2 (4.4%) patients with MYCN gene gain. ALK oncogene testing was performed on 20 patients, and ALK mutations were identified in 5 (25%) patients. One patient was initially diagnosed with a wild-type ALK gene; however, an ALK mutation was detected at the time of disease recurrence. Additionally, 3 of the 12 patients (25%) had ATRX mutations.
In this study, 12 patients underwent WES (5 at initial diagnosis and 7 at disease relapse) to comprehensively characterize the mutational landscape of adolescent and adult patients with NB. The genomic landscape of somatic mutations is shown in Figure 1(a), and the summary of genetic alterations is listed in Supplemental Table S2. Patients were stratified into the response group (CR/PR) and the non-response group (SD/PD) based on their best response to chemotherapy. Our genomic analyses revealed that certain genetic alterations in DNA repair and cell cycle pathways occurred more frequently in the non-response group (SD/PD) than in the response group (CR/PR). Specifically, the mutation rates of ATRX, ARID1B, MLH1, POLD1, TP53, and EED were higher in the PD/SD group than those in the CR/PR group. Representative cases were selected to demonstrate the association between therapeutic response and specific pathway gene mutations. Both Patient 62 and Patient 9 were adults (aged >18 years) diagnosed with poorly differentiated NB, classified as high-risk with INRGSS M stage. Patient 62 exhibited only SD as the best response to chemotherapy, accompanied by mutations in ATRX, ARID1B, and TP53 within DNA repair and cell cycle pathways. In contrast, Patient 9 achieved CR without detectable mutations in these pathways. This divergence in pathway-specific genomic alterations likely contributed to their differential chemosensitivity and clinical outcomes. Following induction therapy, Patient 9 attained CR and received metronomic oral chemotherapy as maintenance treatment. Despite disease recurrence at 18 months, salvage chemotherapy reinduced CR, and the patient has remained disease-free. Conversely, Patient 62 demonstrated poor sensitivity to induction therapy (evaluated as PD), experienced rapid disease progression within 1 month, failed to respond to the same salvage regimen, and succumbed to the disease 4 months post-diagnosis.

(a) The genomic landscape by WES from 12 patients. Clinical information is annotated in the bar plot at the bottom, with each column representing an individual case. Patients were stratified into the response group (CR/PR) and the non-response group (SD/PD) based on their best response to chemotherapy. Genomic analysis revealed a higher prevalence of genetic alterations in DNA repair and cell cycle pathways in the non-response group (SD/PD) compared to the response group (CR/PR). Specifically, mutations in ATRX, ARID1B, MLH1, POLD1, TP53, and EED were more frequently observed in the PD/SD group than in the CR/PR group. (b) Longitudinal monitoring by ctDNA and radiographic evaluation. Swimmer plot showing ctDNA status and clinical response evaluated by imaging. Solid blue dots indicate ctDNA positivity, with color intensity reflecting changes in ctDNA abundance, while hollow dots represent ctDNA negativity.
In addition, six patients underwent plasma ctDNA surveillance, three at initial diagnosis and three at disease progression, as depicted in Figure 1(b). The consistency between ctDNA surveillance and imaging response evaluations was 83.3% (5/6) by comparing the dynamics trend of ctDNA and the disease status around the same time. Among 13 CR events identified by imaging, 61.5% (8/13) exhibited corresponding ctDNA negativity. Patient 30, a high-risk relapse case, initially achieved imaging-confirmed CR but with concurrent ctDNA positivity. Despite two subsequent imaging-confirmed CR assessments, ctDNA remained continuously positive, and disease progression was ultimately detected at the third evaluation, indicating that ctDNA monitoring provided a ⩾4-month lead time for progression prediction. Following treatment, ctDNA clearance paralleled the achievement of sustained CR. Patient 18, a newly diagnosed case, achieved postoperative CR with concordant ctDNA negativity throughout follow-up, demonstrating 100% consistency with imaging assessments. Similarly, Patient 28, a relapsed case, exhibited progressive ctDNA burden reduction following salvage therapy and surgery, culminating in dual CR confirmation of imaging and ctDNA negativity. These cases indicate that ctDNA surveillance demonstrates high concordance with imaging-based response evaluations and enables earlier prediction of disease progression, highlighting its potential as a complementary biomarker for precision monitoring in adolescent and adult NB management.
Treatment and outcomes
All patients underwent chemotherapy (n = 11, 16.9%), surgery (n = 12, 18.5%), or combination induction therapy (n = 42, 64.6%), with or without ASCT (n = 2, 3.1%), or radiation therapy (n = 23, 35.4%). At the end of the first induction therapy, 34 patients (53.2%) achieved overall CR. Other responses included 6 patients with PR, 1 patient with MR, 16 patients with SD, and 8 patients with PD.
Very low-risk and low-risk groups of NB
All 17 patients in the very low-risk and low-risk groups underwent surgical procedures, and complete resection was achieved in 13 (76.5%) patients. Additionally, 6 (35.3%) patients were administered chemotherapy. Following therapy, 16 patients achieved CR, and 1 patient had SD.
Tumor recurrence was observed in four patients, and various salvage treatments were explored, such as chemotherapy regimens for high-risk patients and a second surgery. At the data cutoff date, 1 patient had succumbed to the disease, 3 lived with the tumor, and the remaining 13 patients were disease-free.
Intermediate-risk group of NB
Among the five patients in the intermediate-risk group, four underwent surgery followed by chemotherapy, and one patient received only chemotherapy, with a median of 7 (IQR: 5–8) courses of chemotherapy. Additionally, radiotherapy was administered to two (45%) patients. At the end of treatment, three patients achieved CR, and two had SD.
Tumor recurrence occurred in one patient after 32.5 months, affecting the bones, lymph nodes, and pleura. The patient received subsequent treatments, including surgery, CAV/VIP chemotherapy regimens, VIT (vincristine, irinotecan, temozolomide) regimens, and radiotherapy. At the latest follow-up, one patient had died of disease progression, two were disease-free, and two continued to live with the tumor.
High-risk group of NB
All 42 high-risk patients received dose–intensity induction chemotherapy with a median of 7 (IQR: 5–8) cycles. Surgical intervention was performed in 32 (76.2%) patients, of whom 19 achieved complete resection, 10 underwent partial resection, and 3 lacked detailed surgical records. After induction therapy, 14 (33.3%) patients achieved CR, 6 (14.3%) achieved PR, 1 (2.4%) showed MR, 13 (31%) had SD, and 8 (19%) had PD. In the consolidation therapy phase, 21 (50.0%) patients received radiation therapy: 13 for the primary lesion, 5 for both primary and metastatic lesions, 3 without radiation details, and 2 patients underwent ASCT. Subsequently, 18 (42.9%) patients received maintenance therapy, including two with anti-GD2 antibody (dinutuximab-β and naxitamab), 12 with metronomic chemotherapy, and four with cis-retinoic acid.
Nine patients had disease relapse, 22 had disease progression after first-line multimodal therapy, and the most common sites of recurrence or progression were the primary lesion (n = 11), bone (n = 10), and liver (n = 5). Based on the adopted treatment strategies, molecular testing results, and locations of tumor recurrence, various individualized salvage treatment regimens were attempted. These salvage strategies included IT (irinotecan, temozolomide)-based regimens, CT regimen (cyclophosphamide, topotecan), IC regimen (ifosfamide, carboplatin), and potentially beneficial drugs such as tyrosine kinase inhibitors (TKI: lorlatinib and anlotinib), apatinib, PD1 inhibitors, and chidamide. Second-line chemotherapy based on the IT regimen was administered to 17 patients, resulting in an objective response rate (ORR) of 41.2% (3 CR + 4 PR). Subsequent treatments included surgery in seven patients, radiotherapy in eight patients, anti-GD2 (naxitamab) immunotherapy in one patient, and metronomic chemotherapy in seven patients. Adverse effects in patients who received anti-GD2 immunotherapy were tolerable and manageable, including pain, paresthesia, elevated aminotransferase levels, and blurred vision. At the last follow-up, 18 patients remained alive, 5 were disease-free, and the others continued to live with the tumor. The 5-year PFS and OS rates of high-risk group patients were 16.6% ± 7.1% and 46.6% ± 9.6%, respectively.
Prognostic factors for survival
At a median follow-up time of 53.5 months (IQR: 24.3–86.9), the 3-year PFS and OS rates of the entire cohort were 47.2% ± 6.7% and 78% ± 5.7%, respectively; the 5-year PFS and OS rates were 33.1% ± 6.9% and 55.1% ± 7.6%, respectively (Figure 2).

OS (orange line) and PFS (green line) for the entire cohort. The 3-year PFS and OS rates were 47.2% ± 6.7% and 78% ± 5.7%, respectively; the 5-year PFS and OS rates were 33.1% ± 6.9% and 55.1% ± 7.6%, respectively.
Among the 65 patients, Cox regression was used to assess the prognostic factors for PFS and OS with available follow-up data (n = 60; Table 2). In the univariate analysis, patients in the non-high-risk group (p = 0.001 for PFS, p = 0.011 for OS), locoregional (INRGSS L1 and L2 stage) NB (p = 0.001 for PFS, p = 0.041 for OS), and those who achieved CR after induction therapy (p = 0.003; PFS, p = 0.004 for OS) were associated with better outcomes. In addition, complete tumor resection (p = 0.005) and patients with histologic GNB (p = 0.017) showed superior PFS.
Univariate analysis for survival.

CI, confidence interval; CR, complete response; GNB, ganglionic neuroblastoma; HR, hazard ratio; INRGSS, International Neuroblastoma Risk Group Staging System; NB, neuroblastoma; OS, overall survival; PFS, progression-free survival.
In multivariate analysis, CR after induction therapy (hazard ratio: 3.469, 95% confidence interval (CI): 1.359–8.854, p = 0.009) was independently associated with favorable PFS compared with non-CR (Figure 3). The 5-year PFS of these patients with CR was 44.0% ± 10.6%, significantly superior to that of non-CR patients (18.5% ± 8.8%).

In multivariate analysis, achieving CR after induction therapy was associated with favorable PFS. The 5-year PFS of these patients with CR was 44.0% ± 10.6%, significantly superior to that of non-CR patients (18.5% ± 8.8%).
Comparison between adolescent and adult NB
The clinical and biological characteristics of the adolescent and adult patients with NB had no significant difference (Table 3). The survival data of the 3- and 5-year PFS rates were 41.3% ± 10.9% and 28.3% ± 10.7%, respectively, in adolescent patients; in adult patients, these rates were 55.5% ± 8.7% and 35.3% ± 9.8%, respectively (p = 0.40). The 3- and 5-year OS were 80.9% ± 8.6% and 54.5% ± 11.3% in the adolescent group, respectively; the 3- and 5-year OS were 84.0% ± 6.7% and 52.3% ± 10.7% in adult patients, respectively (p = 0.208). No significant difference was observed between the two groups.
Comparison of clinical, biological characteristics, and prognosis between adolescent and adult neuroblastomas.

INRG, International Neuroblastoma Risk Group; INRGSS, International Neuroblastoma Risk Group Staging System; OS, overall survival; PFS, progression-free survival; NA, not available.
Discussion
NB is rare in adolescents and adults, with unique clinical and biological characteristics. Standard treatment guidelines or chemotherapy protocols based on these populations are lacking, and recommendations are based on the experience from NB in younger patients. Enhancing our empirical understanding of this disease will provide guidance for future clinical practice. Therefore, we conducted a retrospective study to investigate the clinical characteristics, treatment modalities, and prognostic data of this rare disease, with the aim of providing more comprehensive references for clinical practice.
In our study, the observations on clinical characteristics are similar to those reported in previous adolescent and adult series.6–8,17,23 The most common primary site of origin was the adrenal gland, but the most common metastatic site was the lymph nodes, which differed from the patterns in pediatric NB in which more bone marrow involvement was observed. 24 Previous studies have reported unusual patterns of metastatic localization in adolescents and adult patients with NB, such as in the lungs or central nervous system.12,14 Similar to the findings of the pediatric cohort study, 1 patients in the high-risk NB group constituted the highest proportion. However, a higher proportion (61.5%) of patients presented with metastatic disease at diagnosis was observed, with only 40%–55% of pediatric patients presented with Stage 4 disease at diagnosis.25–28 This discrepancy may be attributed to delayed diagnosis due to the rarity of adult-onset NB, as a longer interval between symptom onset and diagnosis has been reported. 7
Although the induction regimens vary across different cooperative groups, the ORR for induction regimens typically ranges from 71% to 85% in pediatric patients with NB.29,30 In contrast, our study showed that adolescent and adult patients with NB had a lower ORR of 61.5% to the first-line induction therapy. Similarly, regarding salvage chemotherapy regimens, a retrospective analysis of 46 pediatric patients with relapsed/refractory NB treated with the VIT regimen at our center demonstrated an ORR of 69.6%. 31 However, the adolescent and adult patients in our study responded poorly to salvage IT-based chemotherapy, with an ORR of only 41.2%. These findings suggest that adolescent and adult patients with NB may have lower chemotherapy sensitivity than pediatric patients, which may contribute to their poorer prognosis. Nevertheless, our multivariate analysis indicated that achieving CR after induction therapy was the only factor associated with better outcomes. Therefore, in clinical practice, an aggressive strategy and complete surgical resection should be adopted to achieve CR after first-line induction therapy to improve the prognosis of adolescent and adult patients with NB.17,32
Age at diagnosis is an independent factor highly correlated with NB prognosis. 33 However, in our subgroup analysis of adolescent and adult patients with NB, we did not observe a significant age-related impact on the outcomes. This aligns with findings from the SEER report 8 that age stratification did not show statistically significant prognostic differences. Nonetheless, when comparing the two major groups of adolescent/adult, and pediatric patients, adolescent and adult patients tend to exhibit poorer prognoses, as previously reported.7,8,10,17 Specifically, in cases of MYCN-nonamplified in pediatric NB, the 3-year OS for the non-high-risk group typically exceeds 95%.34–36 For high-risk pediatric patients, the 5-year OS rate is approximately 62.5%, 37 which is better than the survival rate observed in adults. Despite this, the survival outcomes in our study were more favorable than those of other large-scale studies on adult NB. In our cohort, the 3- and 5-year OS rates for patients over 20 years of age were 84.1% and 63.5%, respectively, which are higher than those of 125 participants based on SEER data, with 3- and 5-year OS rates of 45.9% and 36.3%. 8 Additionally, for adult patients with M-stage disease, the 3-, 5-, and 10-year OS rates were 77.9%, 49.5%, and 24.7%, respectively, which were superior to those reported by Conter et al., 23 in which the 3-, 5-, and 10-year OS rates of 18 patients with M-stage disease were 68%, 33%, and 13%. In the near absence of ASCT and anti-GD2 antibody immunotherapy, our prognosis is comparable to that of a study conducted at the Memorial Sloan Kettering Cancer Center, where 6 of 33 high-risk patients with NB underwent ASCT, and 7 of 33 patients received GD2 antibody treatment. 17 Compared to data from other upper-middle-income countries, our 5-year OS was significantly higher, at 55.1% versus 41.6%, 20.9%, and 14.2% reported by the other three centers.12,38,39
Several factors may have contributed to this discrepancy. Firstly, aggressive and personalized salvage treatment strategies were employed in patients with relapsed or refractory NB. Based on the patient’s individual condition, molecular testing results, and family economic status, various treatment options were employed, including TKIs, VIT regimens, PD-1 monoclonal antibodies, anti-angiogenic drugs, and histone deacetylase inhibitors. Currently, the IT-based regimen has become the backbone chemotherapy for relapsed/refractory pediatric patients with NB, with ORR ranging from 26% to 69.6% when combined with other agents.31,40–43 However, data on its efficacy in adult patients are limited. Our exploratory experience, which showed an ORR of 41.2%, suggests that the IT regimen may serve as an effective salvage therapy for adolescent and adult patients with NB. Moreover, for some high-risk patients, low-dose metronomic maintenance therapy was implemented. Previous studies have indicated that oral metronomic maintenance therapy can improve survival rates in high-risk pediatric patients with NB who have not undergone ASCT or anti-GD2 antibody treatment. 44 Owing to the high cost of GD2 antibodies and the significant toxicity associated with ASCT, these treatments are less accessible in developing countries. Therefore, low-dose metronomic maintenance therapy is a viable alternative. Our limited data also suggest that metronomic maintenance therapy can benefit adult patients with NB. However, further prospective clinical trials are still needed.
NB relatively lacks recurrent somatic mutations, with only a few genes showing significant somatic mutation frequencies, including ALK, PTPN11, ATRX, MYCN, and NRAS. 45 Compared with pediatric patients with NB, adolescent and adult patients exhibit different patterns of genomic aberrations. MYCN amplification has been shown to be associated with advanced tumor stage and disease progression and is used as a biomarker for risk stratification.46,47 MYCN amplification in NB is usually reported in 20%–30% of pediatric patients. 48 However, consistent with other studies,8,10,17 only three patients in our cohort exhibited MYCN amplification. Franks et al. 10 inferred that the lack of MYCN amplification may contribute to the indolent course and slower progression of NB in adults. Conversely, a higher proportion of ALK gene mutations was observed in adult patients with NB, which is consistent with other studies. 17 Genetic alterations in ALK mutations and amplifications have also been observed in approximately 14% of high-risk NB cases and are regarded as independent predictors of poor survival.49,50
However, the genomic landscape of adult NB remains unclear. In our analysis of the genomic landscape of the 12 patients, those with poor responses to chemotherapy had a higher frequency of mutations in the cell cycle and DNA repair pathways. Chemotherapeutic medicines target proliferating cells, and alterations in cell cycle regulation and DNA damage repair can affect chemotherapy. 51 Studies on other cancer types have shown that the cell cycle and DNA repair pathways are associated with tumor sensitivity to chemotherapy.52,53 Further investigation is needed to clarify the functional outcomes associated with these pathways in relation to chemotherapy response. Given the limited available guidelines and poor outcomes observed in adolescent and adult NB patients, comprehensive molecular testing and earlier implementation of targeted therapies may improve clinical outcomes and deepen our understanding of the genomic landscape of this rare malignancy.
ctDNA has been reported to correlate with disease burden, treatment response, and predict relapse.54–56 In our study, the abundance of ctDNA was consistent with treatment response assessments based on imaging. Additionally, the noninvasive nature of ctDNA allows for multiple longitudinal tests. In our study, one patient was initially diagnosed with a wild-type ALK gene, but an ALK gene mutation was detected at recurrence. This reflects the heterogeneity and clonal evolution of tumors. Malignant clones of NB emerge early during embryonic development and rapidly evolve and diversify. 57 Capturing this dynamic and rapid clonal evolution is crucial for the personalized management and treatment of patients with NB. In summary, our limited data suggest that the dynamic monitoring of ctDNA could benefit the management of adult patients with NB, offering a more comprehensive option than single-molecule testing.
This study had some limitations. First, this retrospective study spanned 17 years, resulting in missing medical data. Second, GD2 antibody therapy was approved in China in 2021 and is costly, leading to limited data on anti-GD2 antibody therapy at our center. Finally, only 12 patients underwent genetic molecular testing, restricting a comprehensive molecular understanding of this rare disease.
Conclusion
Compared to pediatric patients, adolescent and adult patients with NB exhibit distinct biological characteristics and poorer prognoses, possibly due to reduced chemotherapy sensitivity. Mutations in the cell cycle and DNA repair pathways are associated with sensitivity to chemotherapy. ctDNA surveillance has potential benefits in the management of adolescent and adult patients with NB. Patients who achieve a CR after induction therapy may have a better prognosis. Metronomic maintenance therapy or various aggressive salvage strategies may improve survival in patients who do not receive anti-GD2 antibody therapy or ASCT. However, further exploration of treatment options is required.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251337494 – Supplemental material for Characteristics, treatments, and outcomes of adolescents and adults with neuroblastoma: a retrospective study in China
Supplemental material, sj-docx-1-tam-10.1177_17588359251337494 for Characteristics, treatments, and outcomes of adolescents and adults with neuroblastoma: a retrospective study in China by Weiji Xie, Yu Zhang, Jiaqian Xu, Feifei Sun, Jia Zhu, Yi Que, Junting Huang, Zijun Zhen, Suying Lu, Juan Wang and Yizhuo Zhang in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251337494 – Supplemental material for Characteristics, treatments, and outcomes of adolescents and adults with neuroblastoma: a retrospective study in China
Supplemental material, sj-docx-2-tam-10.1177_17588359251337494 for Characteristics, treatments, and outcomes of adolescents and adults with neuroblastoma: a retrospective study in China by Weiji Xie, Yu Zhang, Jiaqian Xu, Feifei Sun, Jia Zhu, Yi Que, Junting Huang, Zijun Zhen, Suying Lu, Juan Wang and Yizhuo Zhang in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.