
Abstract
In recent years, several global phase III trials have shown that combinations of immune checkpoint inhibitors (ICIs) offer superior efficacy and survival compared to multi-kinase inhibitors, establishing them as the gold standard for treating patients with advanced hepatocellular carcinoma (HCC). This success has led to investigations into expanding the use of immunotherapy into various other settings and populations, including neoadjuvant and adjuvant therapies, patients with decompensated liver function and those awaiting liver transplantation. Despite its proven efficacy, a significant number of patients still develop resistance to immunotherapy, highlighting the need for innovative strategies to address this challenge. Approaches aimed at enhancing tumour immunogenicity, such as combining immunotherapy with transarterial chemoembolization or radiation therapies, show significant promise. Additionally, novel immunotherapeutics – such as triplet therapy, bispecific antibodies, adoptive T-cell therapy and cancer vaccines – are in early development for HCC. These agents have demonstrated potential for synergistic effects with existing ICIs, with initial studies yielding positive outcomes. In this review, we offer our future perspective on immunotherapy, emphasizing emerging indications, novel combination strategies and the development of new immunotherapeutic agents. Overall, the future of immunotherapy in HCC is brimming with extraordinary potential, set to transform the treatment landscape and redefine the possibilities for managing this challenging disease.
Keywords
Introduction
Systemic treatment for hepatocellular carcinoma (HCC) has made significant advancements over the past decade. The introduction of immune checkpoint inhibitors (ICI) targeting programme death-ligand 1 (PD-(L)1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) has significantly increased survival rates for patients with advanced HCC, extending life expectancy from a few months to over 2 years.1–5 Even more encouraging is that studies have shown up to 25% of patients may survive beyond 4 years with ICI-based treatments – a finding that was much less observed during the era of multi-kinase inhibitors (MKIs). 4 Given these outcomes, ICI-based treatment is now the standard first-line therapy for patients with advanced HCC.6,7
It has long been recognized that the immune system plays a substantial role in cancer control. This observation dates back over a century ago to when bone sarcoma surgeon William Bradley Coley started injecting patients with Coley’s Toxin to stimulate their immune systems to combat cancer. 8 However, it has only become clear in the last 10–20 years how we can harness the power of our immune system to tackle cancer cells. The concept of immunoediting emerged in the early 21st century, emphasizing the role of immunosurveillance in tumour elimination and the immunologic sculpting that occurs during cancer development. 9 The cancer-immunity cycle, introduced in 2013, established a framework and seven key steps necessary for effective immuno-clearance of cancer cells. These include the release of cancer antigens, activation of antigen-presenting cells and T cells, trafficking and infiltration of immune cells into the tumour microenvironment (TME) and effective destruction of cancer cells by the immune system. 10 A recent update further highlights the importance of T-cell migration through the tumour stroma and its complex interactions with intratumuoral immune cells. 11
The enhanced understanding of the relationship between cancer and the immune system, along with the success that ICIs in treating HCC, has sparked significant interest in developing new immunotherapy indications, strategies and agents for HCC treatment. In this review, we offer a forward-looking perspective on immunotherapy for HCC management, focusing on expanding its indication to special populations (such as patients with Child-Pugh (CP) B liver function or those eligible for liver transplantation) and settings (such as neoadjuvant or adjuvant therapy). We also discuss improving immunogenicity through novel combination strategies (e.g. locoregional treatments) and the rise of new immunotherapeutics (e.g. novel ICIs, adoptive cell therapy, bispecific antibodies (BsAb) and therapeutic cancer vaccines; Figure 1).

The future of immunotherapy would focus on expanding the indication to special populations, enhancing tumour’s immunogenicity and the use of novel immunotherapeutics such as bi-specific antibodies, adoptive cell therapies and therapeutic cancer vaccines.
Expansion of indication
Neoadjuvant and adjuvant immunotherapy
Surgical resection or local ablation is considered a curative option for patients with HCC. However, about 70% of these patients will experience recurrence within 5 years, underscoring the need for effective adjuvant therapy. 12 Recurrence typically follows a bimodal distribution: early recurrence peaks at 1–2 years due to occult micrometastases, while late recurrence, occurring around 4–5 years post-treatment, is linked to de novo tumours related to underlying liver disease. 13 Although adjuvant therapy could potentially eliminate micrometastases and reduce early recurrence, no such therapy has proven effective in global phase III trials.14,15
The IMbrave-050 study compared 1 year of adjuvant atezolizumab plus bevacizumab, an effective therapy for advanced HCC, with active surveillance in high-risk patients after curative surgery or ablation. In the first interim analysis after a median follow-up of 17.4 months, the median recurrence-free survival (RFS) was improved with combination therapy (hazard ratio (HR) 0.72; p = 0.012). The treatment group showed a higher rate of 12-month RFS at 78% compared with 65% in the active surveillance arm. However, this benefit was not maintained in the second interim analysis (HR 0.90, 95% confidence interval (CI): 0.72–1.12). Based on the updated results of IMBrave-050, atezolizumab plus bevacizumab was not supported as an adjuvant therapy. Four other studies (CheckMate-9DX, KEYNOTE-937, EMERALD-2 and JUPITER-04) are ongoing to explore any roles of adjuvant immunotherapy for high-risk patients.
Despite the setbacks seen in the adjuvant setting, interest in neoadjuvant immunotherapy is on the rise. Clinical trials have demonstrated that neoadjuvant immunotherapy can improve survival compared to adjuvant treatments across multiple cancers, including melanoma, lung cancer and breast cancer.16–18 Neoadjuvant immunotherapy leverages the intact tumour in situ as a vaccine, generating a broader spectrum of T-cell antigens compared to adjuvant treatment, facilitating a more robust T-cell activation and immune response. 19
One of the earlier studies on neoadjuvant immunotherapy in HCC investigated the use of anti-PD-1 alone, or in combination with anti-CTLA-4. This randomized phase II study involved 27 patients receiving either nivolumab or nivolumab plus ipilimumab. 20 Patients were given three doses of nivolumab every 2 weeks, plus or minus one concurrent dose of ipilimumab during the first cycle, followed by surgery and adjuvant treatment. Among the 27 treated participants, 20 underwent surgical resection, while 7 had surgeries cancelled due to disease progression, concern about the insufficient liver remnant and prohibitive adhesive disease. Regarding safety, grade 3 or higher treatment-related adverse events (TRAEs) were seen in 43% (6/14) of patients receiving nivolumab plus ipilimumab, compared to 23% (3/13) for those on nivolumab alone. Notably, no patients experienced surgery delay due to AEs. Among the 13 patients on nivolumab monotherapy, 23% (3/13) had a radiological response, and 33% (3/9) exhibited a major pathological response (MPR). By contrast, none of the patients on the combination therapy showed a radiological response, but 27% (3/11) achieved an MPR. Importantly, those with MPR did not experience recurrences after a median follow-up of 2 years, while those without such responses had a median RFS of 24.4 months.
The PRIME-HCC study further explores the safety and tolerability of neoadjuvant nivolumab plus ipilimumab. 21 The study involves a 6-week preoperative immunotherapy regimen, including one dose of ipilimumab (1 mg/kg) every 6 weeks, and two doses of nivolumab (3 mg/kg) every 3 weeks. The primary endpoint focuses on the safety and tolerability of nivolumab plus ipilimumab combination, while secondary endpoints assess objective response rate (ORR) and pathological outcomes on resected specimens. An interim report from 2023 indicated that 25 patients were enrolled, with any grade AEs occurring in 88%, including 24% at grade 3. 22 No grade 4 or 5 AEs were reported. Among 21 patients evaluated for radiological response before surgery, the ORR was 29%, and the disease control rate (DCR) was 95%. One patient with mixed HCC/cholangiocarcinoma developed disease progression. An MPR was observed in 56% of resected specimens, with 38% of patients achieving pathological complete remission. 21
Given the limited number of patients in neoadjuvant studies, a recent systematic review and meta-analysis aimed to summarize outcomes and safety data. 23 The review included 11 studies, though 7 were presented as conference abstracts. Amongst these, three studies investigated neoadjuvant anti-PD-1 plus anti-CTLA-4, three examined neoadjuvant anti-PD-1 monotherapy and six looked at combinations of anti-PD-1 plus anti-VEGF or MKIs. Overall, 51.7%–95.2% of patients proceeded to surgery, with grade 3 or 4 TRAEs occurring in 10%–41.4% of cases. Pathological complete responses (pCR) ranged from 5.9% to 38%, with higher rates observed in those treated with the anti-PD-1 and anti-CTLA-4 combination. However, due to the heterogeneous patient population and therapies, it is challenging to generalize these results. Moreover, long-term survival data are lacking, raising questions about whether higher pCR translates into survival benefits. For example, in the CheckMate-816 study, neoadjuvant nivolumab plus chemotherapy improved event-free survival in resectable lung cancer compared to standard arm chemotherapy alone, from 20.8 to 31.6 months. 17 Furthermore, pCR is predictive of improved survival, which is observed in both the experimental arm and control arm. On the contrary, the KEYNOTE-585 study which evaluated the role of perioperative pembrolizumab with chemotherapy in locally advanced resectable gastric, or gastro-oesophageal adenocarcinoma, the improvement in pCR using combination therapy (12.9% vs 2%) did not translate into survival benefits. 24 Therefore, while neoadjuvant immunotherapy appears to enhance pathological responses for HCC, the underlying tumour biology is likely a key factor in determining whether these responses lead to survival benefits.
A recent retrospective study revealed that neoadjuvant immunotherapy may enable high-risk patients, those typically beyond the standard resection criteria, to achieve margin-negative resections with comparable long-term outcomes to upfront surgeries. 25 This study included 92 patients, with 36 receiving neoadjuvant immunotherapy, primarily anti-PD-1 therapies, either as monotherapy (27.8%), in combination with MKIs (36.1%), or with anti-LAG-3 (16.7%). Notably, 61.1% of these patients who received neoadjuvant immunotherapy were outside standard respectability criteria and exhibited high-risk features for recurrence. The findings revealed that patients undergoing neoadjuvant immunotherapy had similar rates of margin-negative resection and RFS compared to those who underwent upfront surgical resection (44.8 vs 49.3 months, p = 0.66).
Currently, evidence suggests that neoadjuvant immunotherapy can improve pathological responses, though studies indicate that radiological responses may not correlate with these outcomes. There is a pressing need for better biomarkers for response assessment to neoadjuvant immunotherapy for HCC. Additionally, longer follow-up is needed to determine whether neoadjuvant immunotherapy improves overall survival (OS) and whether pathological response can serve as a surrogate endpoint for survival.
At the moment, several early-phase neoadjuvant studies are ongoing, exploring various combinations of neoadjuvant immunotherapy. 26 These include the NEOTOMA trial (NCT05440864), which evaluates neoadjuvant tremelimumab and durvalumab in resectable HCC, a trial assessing the role of LAG-3 inhibitor relatlimab plus nivolumab in potentially resectable HCC (NCT04658147) and a study investigating combinations of neoadjuvant anti-PD-1 plus MKIs (NEO-LEAP (NCT05389527)). These studies will improve our understanding of the role of neoadjuvant immunotherapy and clarify the optimal combination approach, patient selection and endpoints for future phase III studies.
Child-Pugh B population
HCC often develops in patients with cirrhosis, which can lead to liver failure. 27 Consequently, those with CP-B liver function are frequently excluded from registered phase III clinical trials due to poor prognosis and concerns about tolerability. This has resulted in recommendations for the best supportive care for these patients. 6 Data on systemic treatments in patients with CP-B patients are limited, mainly consisting of retrospective studies. Traditionally, a small proportion of patients may receive sorafenib, based on large-scale observation studies that showed modest OS benefits compared to supportive care. 28 A meta-analysis of 30 studies involving 8678 patients with advanced HCC indicated that CP-B patients had worse OS (4.6 months) than those with CP-A (8.8 months), with no significant difference in safety profiles between the groups. 29 Overall survival rates in patients with CP-A were 8.8 months and 4.6 months in CP-B. There was no significant difference in safety and tolerability between the two CP groups, with 35% of patients experiencing grade 3 or 4 adverse events. 29 With regard to lenvatinib, a small number of retrospective studies revealed similar tolerability in CP-B patients, though the relative dose intensity was only 50%–75% of the planned dose. The reported OS is in the range of 6–12 months.30–32
Immunotherapy has significantly improved survival in patients with optimal liver function substantially (ClinicalTrials.gov identifier: NCT04039607).33,34 Although a proportion of patients experience immune-mediated adverse events (imAEs), most can be managed and few patients require discontinuation of treatment.34–36 The favourable safety profile and efficacy of immunotherapy compared to MKIs have led to an interest in its use in patients with suboptimal liver function. The CheckMate-040 trial (cohort 5) was the first prospective study on immunotherapy in CP-B patients. 37 It examined single-agent nivolumab in 49 CP-B patients with advanced HCC. Of note, DCR was 55% and the median OS was 7.6 months, with longer survival seen in sorafenib-naïve patients (median OS: 9.8 months). Notably, 24% experienced grade 3 or 4 TRAEs, but high-grade hepatobiliary disorders were uncommon (n = 3), suggesting nivolumab is generally safe in this population. Several retrospective studies also supported the safety of nivolumab in CP-B patients, with similar OS observed (Table 1).38–40
Outcomes of selected studies on immunotherapy in Child-Pugh B patients.
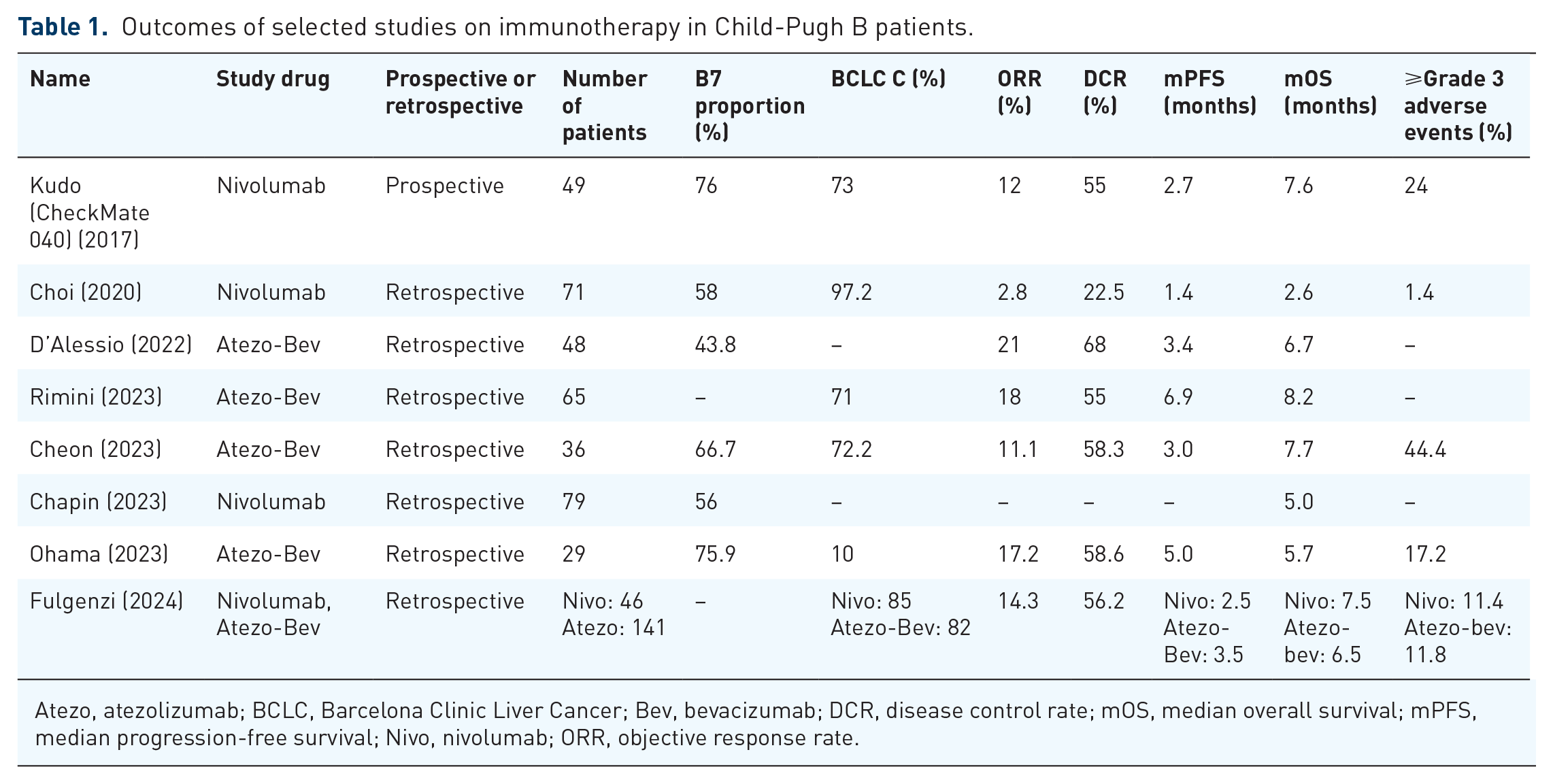
Atezo, atezolizumab; BCLC, Barcelona Clinic Liver Cancer; Bev, bevacizumab; DCR, disease control rate; mOS, median overall survival; mPFS, median progression-free survival; Nivo, nivolumab; ORR, objective response rate.
Additionally, some studies have assessed the safety and efficacy of atezolizumab plus bevacizumab in CP-B patients. The combination is a standard first-line treatment for advanced HCC, demonstrating improved OS compared to sorafenib in the IMbrave-150 study. 33 However, concerns about tolerability, especially bleeding events, in patients with severe cirrhosis, have led to hesitance in using this combination for those with suboptimal liver function. Patients with suboptimal liver function are often complicated with severe portal hypertension, leading to a higher risk of thrombocytopenia, abnormal clotting function and variceal bleeding. An early retrospective study compared the efficacy and safety of atezolizumab plus bevacizumab in 202 patients with CP-A or CP-B liver function, both the median progression-free survival (PFS; 3.4 vs 7.6 months, p = 0.03) and OS (6.7 vs 16.8 months, p = 0.03) were significantly shorter in CP-B patients. 41 While toxicity rates were reported to be similar across both groups, there was a numerically higher incidence of gastrointestinal bleeding in CP-B patients (4% vs 10%). 41 In another retrospective study that explored the use of first-line atezolizumab plus bevacizumab in CP-B patients, the median PFS was 3.5 months and the median OS was 6.5 months. 40 Grade 3 or higher AEs were observed in 11.8% of patients and 2.8% of patients developed grade 3 variceal bleeding (Table 1). 40 Although the studies did not provide the details on the proportions of patients who had undergone pre-treatment oesophago-gastro-duodenoscopy, these figures were slightly higher than the rate of high-grade variceal bleeding reported in the IMbrave-150 study (1%–2%), 33 suggestive of a mild elevation of bleeding risk in CP-B patients.
Another concern with atezolizumab plus bevacizumab is the potential deterioration of liver function over time. A retrospective study involving 147 patients indicated that treatment with atezolizumab plus bevacizumab could lead to deterioration of liver function, manifested as new ascites and hepato-encephalopathy (HE), especially in those with pre-existing liver impairment. 42 Among those meeting the IMbrave-150 inclusion criteria, new ascites and HE were observed in 9.5% and 1.4% of patients, respectively. For patients who were beyond the inclusion criteria of IMbrave-150 (e.g. CP-B7 liver function), a higher proportion of patients experienced liver decompensation with the occurrence of large-volume ascites and high-grade HE requiring hospitalization in 19.2% and 8.2%, respectively. This aligns with the literature suggesting that anti-angiogenic agents like lenvatinib and bevacizumab can impair liver function.43,44 This is because VEGF stimulates the production of nitric oxide (NO) by endothelial NO synthase and increases vascular dilatation and permeability. 45 The use of anti-VEGF may lead to a reduction in NO, resulting in decreased hepatic perfusion and subsequent deterioration of liver function.
Thus, anti-VEGF agents may not be suitable for CP-B patients due to their higher risk of liver decompensation. Nivolumab monotherapy appears to be safe and effective. The SIERRA study is a phase IIIb study exploring the combination of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) in CP-B patients. 46 This study will improve our understanding of whether patients with suboptimal liver function would benefit from the addition of an anti-CTLA-4 agent.
Liver transplant
Liver transplantation is an attractive curative option for patients with HCC, addressing both the cancer and the underlying liver disease. Outcomes of liver transplantation under the Milan criteria are excellent, with an 85%–90% disease-free survival and 70%–80% OS at 5 years.47,48 Importantly, some patients may benefit from transplantation after successful downstaging to within Milan criteria using locoregional therapies, achieving similar 5-year survival rates as those initially meeting the criteria.49,50
The success of combination immunotherapy in advanced HCC has sparked interest in incorporating immunotherapy for earlier-stage HCC or as part of a downstaging protocol before liver transplantation. However, the adoption of immunotherapy in liver transplant patients has been slow due to concerns about graft rejection, which can be fatal.51,52 On the contrary, there were reports on promising outcomes following pretransplant immunotherapy with acceptable safety. 53 In early cohorts involving nivolumab before liver transplantation, no severe allograft rejection was reported, with only one mild acute rejection linked to low tacrolimus level, which responded quickly to increased dosage. 53 The explant pathology revealed over 90% tumour necrosis in one-third of cases, with no recurrences or deaths at a median follow-up of 16 months.
A recent meta-analysis summarized the effects of pretransplant immunotherapy on post-transplant outcomes in HCC. 54 In this study, 91 patients were included, most of whom were beyond Milan criteria (81%) before treatment of immunotherapy and were classified as BCLC B (45.5%). Nivolumab was used in nearly half of the cases (49.5%), either alone or in combination with other immunotherapy. Other common pretransplant immunotherapy regimens included pembrolizumab (23.1%) and atezolizumab plus bevacizumab (15.4%). The study found that young age (adjusted HR per 10 years: 0.72 (95% CI: 0.53–0.99)) and shorter immunotherapy washout periods (adjusted HR per 1 week: 0.92 (95% CI: 0.86–0.99)) were associated with a higher risk of allograft rejection. Based on these findings and also the half-life of anti-PD-1, the authors suggested a 3-month washout may minimize graft rejection risk. After immunotherapy, 11.1% of patients had no viable tumour. Recurrence was observed in nine patients (9.9%), with lower recurrence rates in those within the Milan criteria (16.7% vs 65.3%). Twenty-four patients (26.4%) developed graft rejection but the majority resolved with interventions such as the use of intravenous immunoglobulin or increasing the dose of anti-rejection medication. Overall, the 4-year OS exceeded 80% for the entire cohort, with no significant survival differences between those with and without graft rejection (HR 2.16 (95% CI: 0.58–8.1)).
Current evidence suggests that pretransplant immunotherapy is probably safe, with low graft rejection risk if an appropriate washout period is provided. 55 However, prospective studies are needed to better characterize the safety and efficacy of immunotherapy in patients listed for liver transplantation. While pretransplant immunotherapy should not be routinely offered in clinical practice, enrolling suitable patients into clinical trials testing pretransplant immunotherapy should be considered if available. Several prospective studies are exploring various immunotherapy regimens for downstaging before transplantation, including the ESR-20-21010 evaluating the durvalumab plus tremelimumab in patients who have tumour burden within the UCSF criteria, 56 and the ImmunoXXL study exploring the efficacy and safety of atezolizumab plus bevacizumab in patients with beyond up-to-7 tumour burden. 57
With these studies investigating the efficacy and safety of pretransplant immunotherapy, there remains a major need to understand which high-risk patients with HCC are suitable candidates for pretransplant immunotherapy. Additionally, prospective studies comparing the efficacy and safety of immunotherapy and locoregional therapy as downstaging strategies before liver transplantation are necessary. Expanding immunotherapy indications for liver transplantation could improve access for patients previously deemed ineligible due to tumour burden and enable patients to maintain disease control while awaiting transplantation. 55
Enhance immunogenicity
Combination with transarterial chemoembolization
The BCLC guideline recommends transarterial chemoembolization (TACE) as the standard treatment for patients with intermediate-stage HCC (i.e. multifocal HCC limited to the liver) who are not candidates for curative treatment options like resection, ablation or transplantation. 6 This recommendation arose from the era when there was little effective systemic therapy and patients with more advanced diseases were treated with the best supportive care, while TACE was the only treatment option shown to improve survival compared to supportive care in unresectable HCC.58,59 Consequently, intermediate-stage HCC includes a heterogeneous group of patients with varying tumour burdens, liver function and comorbidities. Although some patients can be effectively treated with one or two sessions of super-selective TACE, repeated TACE can damage the liver function, 60 potentially disqualifying patients from future effective systemic therapies when the disease progresses. Moreover, the median PFS for TACE alone is only about 7–8 months, highlighting a huge unmet clinical need. Therefore, strategies combining TACE with systemic therapies have been explored to enhance anti-tumour response and improve survival. Unfortunately, early studies assessing TACE plus MKIs largely failed, with criticism over study design and endpoint selection.61–63
Recent advances in immunotherapy have prompted investigations into combining TACE with immunotherapeutic agents. It is hypothesized that TACE may prime the TME for immunotherapy and anti-VEGF treatment by releasing neoantigens and inducing ischaemia. 64 The combination of immunotherapy, anti-VEGF therapy, and TACE is thought to bolster anti-tumour responses through immune activation and inhibition of tumour neovascularization. Notably, two recent studies have shown promising results with this combination in TACE-eligible patients with unresectable disease, marking the first evidence of improved responses and survival since TACE was established 20 years ago as a treatment for intermediate-stage HCC.
The EMERALD-1 study is the first phase III trial designed to evaluate the combination of immunotherapy, anti-VEGF and TACE versus TACE alone in patients with HCC not suitable for curative therapy but eligible for embolization. 65 The study randomized 616 patients into three arms. Patients in arm A received durvalumab plus one–four sessions of TACE within the first 16 weeks, followed by durvalumab; those in arm B received durvalumab plus TACE followed by durvalumab and bevacizumab and arm C received TACE plus placebo. The primary endpoint compared the median PFS between arms B and C, with secondary endpoints including PFS between arms A and C, OS, ORR and quality of life. The primary endpoint was met, showing a median PFS of 15.0 months for the combination group compared to 8.2 months for TACE alone (HR 0.77 (95% CI: 0.61–0.98); p = 0.032). Interestingly, the addition of durvalumab did not significantly improve PFS (10 vs 8.2 months; HR 0.94 (95% CI: 0.75–1.19); p = 0.638), suggesting that the PFS benefit was primarily driven by bevacizumab. An absolute 15% increase in ORR was observed in the combination arm (43.6% vs 29.6%, odds ratio: 1.67 (95% CI: 1.10–2.54)). Regarding safety, 45.5% of patients developed grade 3 or 4 toxicity in the combination arm, compared to 23.0% in the TACE alone arm, with hypertension, post-embolization syndrome and hypothyroidism being the most common AEs.
The LEAP-012 is another phase III study evaluating the efficacy and safety of the TACE plus immunotherapy approach. 66 In this global study, 480 patients with intermediate-stage HCC were randomized to receive lenvatinib plus pembrolizumab or a dual placebo in conjunction with TACE. Unlike the EMERALD-1 study, systemic therapy began 2–4 weeks before TACE, with lenvatinib withheld only briefly around the procedure. The primary endpoints were median PFS and OS. At the first interim analysis presented in ESMO Congress 2024, the study showed significant PFS improvement with lenvatinib plus pembrolizumab combined with TACE, compared to TACE alone (14.6 vs 10.0 months; HR 0.66 (95% CI: 0.51–0.84); p = 0.0002). Importantly, there was an early separation of the PFS curves during the period of TACE in the combination arm, further strengthening the hypothesis that the PFS benefits observed in these TACE plus immunotherapy trials were driven by anti-VEGF. The OS data remain immature but suggest a trend favouring the combination arm (HR 0.80 (95% CI: 0.57–1.11); p = 0.0867). ORR was 46.8% in the combination group versus 33.3% in the TACE group, with grade 3 or higher TRAEs occurring in 71.3% of patients in the combination arm and 31.1% for the TACE arm. Treatment-related deaths were reported in 1.7% of patients and 0.4% in the experimental and control arm, respectively.
The results of EMERALD-1 and LEAP-012 establish a new standard for future trials in unresectable HCC patients eligible for embolization. However, it remains uncertain which patient populations should receive this combination approach, as some may be effectively treated with systemic therapy alone, using TACE as salvage therapy. One potential indication from these studies is their ability to downstage tumours for conversion to resectable status, given their higher response rates compared to either systemic treatments or TACE alone (Table 2). The findings of these two studies also enhance our understanding of the potential synergy between TACE and immunotherapy. From the perspective of improving PFS, it appears that combining TACE with single-agent anti-PD-1 is insufficient to elicit a synergistic anti-tumour response, suggesting that anti-VEGF agents play a more critical role. However, it remains to be seen whether a small subset of patients may achieve long-term responses due to enhanced immunogenicity of the combination therapy, a question that future landmark analyses may clarify.
Outcomes of phase III trials evaluating efficacy and safety of immunotherapy in unresectable HCC.

BCLC, Barcelona Clinic Liver Cancer; HCC, hepatocellular carcinoma; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; TACE, transarterial chemoembolization; TRAE, treatment-related adverse events.
Combination with hepatic arterial infusion chemotherapy
Hepatic arterial infusion chemotherapy (HAIC) is a locoregional therapy commonly used in the management of advanced HCC in Asia, particularly in China, Japan and Korea.67–69 HAIC involves the catheterization of the feeding hepatic artery to directly administer chemotherapy, typically platinum-based, to the tumour. This method offers the advantage of delivering a higher dose of chemotherapy directly to the tumour while causing fewer systemic side effects. Recent studies have demonstrated encouraging outcomes with the combination of HAIC and immunotherapy.70–72 In a retrospective study involving 229 patients with advanced HCC treated with either HAIC and immunotherapy (n = 81) or HAIC alone (n = 148), the combination therapy exhibited improved median PFS (10 vs 5.6 months, p = 0.018) and median OS (18.0 vs 14.6 months, p = 0.006). 70 Incorporating HAIC with lenvatinib and anti-PD-1 therapy has also been demonstrated to enhance survival and response rates in patients with advanced HCC. 71 The TRIPLET study, a prospective, single-arm, phase II trial, investigated the efficacy and safety of the combination of HAIC with camrelizumab and rivoceranib in patients with BCLC-C HCC. 72 The triplet treatment showed a high ORR of 71% and a DCR of 87%. The median PFS was 9.4 months and the 1-year OS was 86%. 72 Grade ⩾3 adverse events occurred in 74% of patients, in which the most common AEs were decreased neutrophils and lymphocytes and increased alanine transaminase (ALT) and aspartate transaminase (AST). 72
Combination with radiation-based treatment
Radiotherapy is a promising strategy to sensitize tumours to immunotherapy by delivering precise photon energy to the tumour that causes DNA damage and cancer cell death. The resultant inflammation and release of cancer antigens can increase the immunogenicity within the TME, potentially enhancing the effects of immunotherapy. The phase II START-FIT trial evaluated the sequential use of TACE, stereotactic body radiotherapy (SBRT) and avelumab (anti-PD-L1) in patients with liver-only HCC who were not candidates for curative treatments. 73 In this trial, 33 patients with high burden, liver-limited HCC were recruited, revealing that 55% became amenable to curative treatment, and a 2-year OS of 94% and an ORR by modified RECIST of 67%. 73 Although 33% experienced grade 3 or higher TRAEs, mainly hepatic, this was generally well tolerated. 73
An extension of the study assessed long-term outcomes for patients achieving CR after combined locoregional therapy and immunotherapy. 74 Among 29 patients (46.0%) achieved CR, 3-year time-to-progression was 58.7%, and local control was 90.5%. Among the 10 patients who developed recurrence, 6 of them had solitary intrahepatic disease relapse and subsequently underwent curative surgical treatment. In a phase I study combining SBRT with nivolumab, with or without ipilimumab, those receiving nivolumab plus ipilimumab had an ORR of 57%, median PFS of 11.6 months and OS 41.6 months. 75 In another retrospective series that evaluated the role of SBRT in patients with oligoprogression on immunotherapy, survival was prolonged compared to the historical cohort, even in patients with advanced diseases such as portal vein tumour thrombosis. 76 Despite the small number of patients included, these early results suggest promising synergism between radiotherapy and immunotherapy in advanced HCC. Ongoing studies evaluating the combinations of radiotherapy with various immunotherapies, including atezolizumab plus bevacizumab (NCT06339424, NCT04857684, NCT05992220, NCT06434480), durvalumab (NCT04913480), durvalumab plus tremelimumab (NCT03482102, NCT04430452), toripalimab (NCT04709380) and tislelizumab (NCT05917431).
There is also increasing interest in using Y-90 radioembolization for unresectable HCC. This approach allows for ‘radiation segmentectomy’ of large tumours limited to one liver segment, achieving excellent local response and survival. 77 The promising findings with Y-90 have prompted clinical interest in assessing the combination of Y-90 and immunotherapy. In a phase II study evaluating Y-90 followed by nivolumab, 1 out of 36 patients achieved CR, and 10 achieved PR, according to RECIST 1.1. Median PFS was 5.6 months and OS was 16.9 months. 78 In another phase II study evaluating the combination of pembrolizumab with Y-90 patients with advanced HCC reported an ORR of 30.8%, a median PFS of 10 months and OS of 27.3 months. 79 Several trials are ongoing exploring the combination of Y-90 with different immunotherapy, including atezolizumab plus bevacizumab (NCT05377034), sintilimab (NCT05592584, NCT06397222) and durvalumab plus tremelimumab (NCT04522544).
Overall, emerging evidence suggests that combining radiotherapy with immunotherapy may improve efficacy and survival, even in advanced unresectable HCC. However, longer-term data and larger-scale randomized trials are needed to confirm the OS benefits.
Novel strategies and therapeutics
Using immune modulation to manage HCC is appealing because the liver is intrinsically an immunologically tolerant organ due to the constant efflux of food and infective antigens through the portal venous system. 80 This immunosuppressive environment creates the perfect soil for tumour growth. The transformative success of immunotherapy in HCC treatment largely stems from ICIs targeting PD-(L)1 and CTLA-4. However, there are additional co-stimulatory or co-inhibitory receptors such as TIGIT, LAG-3 and TIM-3, which can be targeted to boost anti-tumour immune response. Furthermore, strategies to overcome T-cell exhaustion – crucial for effective anti-tumour activity – are being actively explored. These strategies may include adoptive cell therapy like CAR-T to enhance T-cell function, bispecific T-cell engagers (BiTEs) or antibodies to improve recruitment of T-cell recruitment and therapeutic cancer vaccines to enhance antigen recognition. 81
Triplets therapy
Anti-TIGIT is one of the major ICIs currently under active investigation. TIGIT is expressed in activated CD8+ T and CD4+ T cells, NK cells and Tregs, but is only weakly expressed in naïve T cells. 82 It can also be expressed on tumour cells, allowing them to evade immune destruction. Its activation suppresses both innate and adaptive immunity through various mechanisms, including dampening T-cell receptor activation signals, inhibiting NK cell degranulation and enhancing the immunosuppressive function of Tregs. 82 Furthermore, in preclinical models, dual blockade of PD-1 and TIGIT has been shown to activate CD226 activation, a costimulatory receptor that promotes TCR signalling and the production of proinflammatory cytokines. 82
The Morpheus-Liver study was a multi-cohort phase Ib/II randomized study comparing the triple therapy of atezolizumab, bevacizumab and tiragolumab (anti-TIGIT), with atezolizumab plus bevacizumab. 83 In the study, 58 patients were randomized, with 40 in the triplet arm and 18 in the doublet arm. The ORR was 42.5% in the triplet group compared to 11.1% in the control group, and the median PFS improved to 11 months from 4.2 months. Responses and survival were observed independent of PD-L1 status. Safety profiles were similar between the groups, with grade 3 or 4 TRAEs occurring in about 30% and AEs leading to discontinuation in 22%. 83 Based on these results, a phase III trial, IMbrave-152, is currently underway. 84
Anti-LAG-3 is another promising ICI in cancer immunotherapy. 85 LAG-3 is an inhibitory receptor highly expressed by exhausted T cells. Its expression negatively regulates T-cell proliferation and effector T-cell function. It binds strongly to class II MHC molecules, 86 disrupting the interaction between co-receptors CD4 and CD8 and the tyrosine kinase Lck, thereby suppressing TCR signalling and T-cell activation. 87 LAG-3 and PD-1 are distinct inhibitory checkpoints often co-expressed on tumour-infiltrating lymphocytes, contributing to tumour-mediated T-cell exhaustion. 88 Preclinical models suggest that LAG-3 expression on CD4+ T cells may limit the effectiveness of anti-PD-1 therapies, supporting the rationale for combination therapy with anti-LAG-3 and anti-PD-1. 89
Relatlimab is a first-in-class anti-LAG-3 antibody that has been shown to restore effective T-cell function. It is currently being tested in various trials for different cancers.90–92 In melanoma, the combination of relatlimab and nivolumab has demonstrated improved survival compared to nivolumab alone, 88 leading to its first FDA approval for clinical use. 86 In HCC, the phase I/II RELATIVITY-106 study is investigating the combination of nivolumab, bevacizumab and relatlimab as a first-line treatment for advanced HCC, comparing the triplet with nivolumab plus bevacizumab.
Another triplet combination of interest involves anti-PD-(L)1, anti-VEGF and anti-CTLA-4, the three most effective immunotherapies for HCC to date. The TRIPLET-HCC study (NCT05665348) is a French randomized phase II/III trial designed to evaluate the efficacy and safety of atezolizumab, bevacizumab and ipilimumab (1 mg/kg for the first four cycles) versus the doublet therapy with atezolizumab and bevacizumab in HCC patients eligible for first-line systemic therapy. 93 A major concern with repeated anti-CTLA-4 dosing is the potential for increased toxicity. Preliminary safety data from the trial included an analysis of 15 patients in the triplet arm, revealing two deaths, with one possibly linked to immune-related hepatitis associated with sepsis, and the other of known cause. 94 Grade 3 or 4 TRAEs were reported in 20% of patients (n = 3). Given the acceptable safety, the trial has decided to continue recruitment.
Bispecific antibodies
BsAb has garnered significant interest for its ability to bind two different epitopes simultaneously, bringing them close together. 95 The dual targeting of BsAbs can work in trans by linking different cells, or in cis by bringing receptors on the same cells into proximity, enhancing receptor signalling. 95
A notable application of BsAbs in HCC is the combination of two ICIs. The blockade of CTLA-4 and PD-(L)1 has shown improved survival rates in HCC, but the therapeutic dosage is often limited by anti-CTLA-4-related imAEs. 96 Interestingly, preclinical studies suggest that local administration of anti-CTLA-4 can effectively mediate anti-tumour activity while minimizing peripheral immune activation, supporting the rationale for developing BsAbs targeting both CTLA-4 and PD-(L)1 in the TME. 97
Cadonilimab, a first-in-class, humanized BsAb targeting PD-1 and CTLA-4, features an Fc null design to enhance safety by reducing antibody-dependent cellular cytotoxicity. 98 Furthermore, it has a higher binding avidity in areas where antigens PD-1 and CTLA-4 are co-expressed, such as in the TME, resulting in reduced peripheral exposure. 99 Cadonilimab was evaluated in two dosing strategies with lenvatinib in phase II trials in patients with advanced. In cohort A (n = 31) in which cadonilimab was dosed at 6 mg/kg every 2 weeks, the combination demonstrated an ORR of 35.5%, median PFS of 8.6 months, and median OS of 27.1 months. In cohort B (n = 28) in which cadonilimab was given at 15 mg/kg every 3 weeks, the ORR was 35.7%, the median PFS was 9.8 months and the median OS was not reached. Overall, grade ⩾3 TRAEs occurred in 66.1% of patients. The most common TRAEs included thrombocytopenia, proteinuria and hypertension, which were consistent with the known AEs related to lenvatinib. 99 Another phase II trial evaluated another PD-1/CTLA-4 BsAbs (AK104) plus lenvatinib showed a similar ORR at 44.4%. Grade 3 or higher TRAEs occurred in 26.7% of patients. 100
Volrustomig, previously known as MEDI5752, is another novel BsAbs targeting CTLA-4 and PD-1, designed to preferentially inhibit CTLA-4 on activated PD-1+ T cells. 96 Early studies suggest it can enhance immune activation with lower toxicity compared to traditional monoclonal antibody combinations. 96 The ongoing GEMINI-hepatobiliary study is evaluating volrustomig’s efficacy and safety as monotherapy and in combination with bevacizumab or lenvatinib. 101 Another study is ongoing evaluating a BsAbs targeting PD-1/LAG-3 co-inhibition. 102
BsAbs can also activate anti-tumour immunity by redirecting immune cells to tumour cells, a strategy known as BiTEs. 103 These typically consist of two single-chain variable fragments (scFv), that bind to tumour cells and CD3 on T cells. 104 While promising results have emerged in other cancers, data in HCC remain limited and is restricted to preclinical models thus far. 105 For instance, a glypican-3 (GPC3)/CD3 BiTE has shown tumour regression in mouse models, 106 and a bispecific NK-cell engager targeting CD16A and GPC3 has demonstrated activity in xenograft tumour models. 107 It would be interesting to see whether these BiTEs will be further developed and tested in clinical trials.
Adoptive cell therapies
Chimeric antigen receptor T-cell therapy (CAR-T) represents one of the most promising strategies for resolving the major limitations of current immunotherapies, which are largely dominated by ICIs. Unlike ICIs, which primarily aim to restore the body’s effective immune responses, CAR-T therapy is a form of ‘synthetic immunity’. It relies on engineering T cells to carry specialized receptors – known as CARs – on their surface. These receptors enable T cells to identify and kill tumour cells expressing specific antigens, independent of MHC presentation.108,109 Because of that, CAR-T therapy can be more potent than other T-cell-based therapies like TCR-T, which depend on the presence of MHC-bound peptides for T-cell activation.
The design of CAR-T is critical to its function, and typically consists of (1) an extracellular domain, an antibody-derived single-chain variable fragments (scFv) antigen-binding domain, (2) a transmembrane domain and (3) an intracellular signalling domain which incorporates signal transduction property and mediates T-cell activation. 110 Upon binding to its target antigen, the CAR triggers T-cell activation. Due to the tissue distribution of cancer cells in haematological malignancies, CAR-T has become one of the most impactful therapeutic strategies for haematological malignancies in the last decade, leading to the approval of CD19-targeting CAR-T for relapsed and refractory B-cell malignancy. 111 However, progress on CAR-T for solid cancer has been slower, largely due to the difficulty of identifying tumour antigens without causing significant off-target effects.
Advancement in CAR-T design employing ‘multi-antigen logic gates’, which restrict T-cell activation to situations that meet multiple conditions, is a promising strategy to overcome the hurdle of on-target, off-tumour toxicity. For example, in the ‘IF-THEN’ strategy, binding to antigen 1 triggers intracellular signalling that induces expression of a separate CAR targeting antigen 2, thereby requiring recognition of both antigens before full T-cell activation. 109 One well-known example is the Syn-Notch system, which uses a synthetic Notch receptor (the first receptor) to drive transcription of a second CAR upon antigen encounter. 112 In glioblastoma (GBM), the epidermal growth factor receptor splice variant III (EGFRvIII) is a highly specific neoantigen found in a subset of GBM cells, making it a perfect target for CAR development. However, an early phase trial evaluating anti-EGFRvIII CAR did not result in long-term survival due to the presence of EGFRvIII mosaism, antigen loss and adaptive resistance. 113 To address this, researchers developed a syn-Notch CAR that first targets the heterogeneously expressed, tumour-specific EGFRvIII, which induces the expression of a second CAR that targets the more homogeneously expressed, but imperfectly tumour-specific antigens (TSA) such as EphA2 or IL13Ra2. This approach has shown strong anti-tumour effects in a mouse model without an off-tumour effect, and a phase I trial (NCT06186401) is now underway. 114
Another strategy for enhancing CAR-T function is to neutralize the immunosuppressive signals within the TME. For example, the transforming growth factor-β (TGFβ) helps the tumour evade immune attack by suppressing T-cell function and promoting tumour progression. A dominant-negative TGFβ receptor – engineered to bind TGFβ but lacking the intracellular signalling portion – can be integrated into CAR-T cells to block inhibitory TGFβ signal, thereby preserving T-cell function.
For HCC, one promising CAR-T target antigen is GPC3. GPC3 is a protein that is found on the placenta and plays a role in morphogenesis. It has been identified to be expressed in several solid tumours, including around 70% of cases of HCC, which is an indicator of poor prognosis. 115 Of note, GPC3 is minimally expressed in normal tissues and cirrhotic liver, making it an attractive target for further development in cell therapy. In a phase I study that evaluates a GPC3-specific TGFRβ armoured autologous CAR-T therapy (C-CAR031) in patients with unresectable HBV-related HCC who were heavily pretreated with systemic therapies (median ⩾3 prior lines), 24 patients were infused with C-CAR031 at various dose levels. 116 Grade 3 or 4 toxicity was experienced in nine (37.5%) of patients. Of interest, the majority of patients experienced low-grade cytokine release syndrome (CRS), with only one patient developing grade 3 CRS. None of the patients developed immune effector cell-associated neurotoxicity syndrome. Despite the small number of patients, the response rates were encouraging with ORR at 56.5% and DCR at 91.3%. The median duration of response was 7.4 months. A translational study indicated that there was a rapid expansion of CAR-T cells and a significant drop in AFP following CAR infusion.
Cancer vaccines
Therapeutic cancer vaccines are designed to activate the immune system to recognize antigens presented on cancer cells, triggering an anti-tumour immune response similar to that against infections. Cancer antigens can generally be categorized into tumour-associated antigens (TAA), TSA and cancer/testis antigens (CTA). 117 TAAs are expressed at low levels in normal cells but at disproportionately higher levels in tumour cells. These antigens often result from genetic amplifications or post-translational modifications. Examples include ERBB2 in breast cancer and CD19 in B-cell malignancies. By contrast, TSAs are completely absent from normal cells and are unique to cancer. They typically arise from oncogenic viral proteins, or non-synonymous somatic mutations, such as HPV oncoproteins E6 and E7 or specific KRAS mutations. CTAs are generally not found in adult cells, except in reproductive tissues, but can be selectively expressed by various tumour types; examples include MAGE and NY-ESO-1. 117
Most cancer vaccine development has focused on widely overexpressed TAAs, which can be produced off-the-shelf, and potentially benefit many patients with the same vaccine. However, many attempts have met with limited success due to the presence of these antigens in normal cells, resulting in their low immunogenicity caused by central and peripheral tolerance mechanisms. 118 For example, a phase II trial of GV1001, which utilized HCC-associated antigens with telomerase-derived peptide in 40 patients with advanced HCC, found no objective responses or specific immune responses following vaccination. 119
Advancements in tumour molecular sequencing technologies have accelerated the development of personalized vaccines that target TSAs. Neoantigens can now be predicted in silico, using whole-exome sequencing to map the tumour-specific mutanome, with RNA sequencing incorporating alternative splicing events. 120 Predicted neoantigens undergo experimental verifications for epitope validation to ensure their immunogenicity. 120 This approach is more efficient than traditional methods, which require cloning tumour antigens and cultivating tumour cell lines. The emergence of mRNA-based vaccines allows for the simultaneous encoding of multiple antigens, generally offering greater immunogenicity than DNA and peptide vaccines, with a lower risk of host genome integration. 121
The clinical efficacy of therapeutic cancer vaccines for HCC is still in the nascent stage, but emerging data suggest augmented efficacy when combined with anti-PD-1 checkpoint inhibitors. The KEYNOTE-942 trial was the first randomized trial to demonstrate a clinically significant benefit from an individualized neoantigen vaccine approach. 122 In this phase IIb trial, 157 patients with completely resected stage IIIB–IV melanoma received either an adjuvant personalized mRNA vaccine with pembrolizumab or pembrolizumab alone. At a median follow-up of 2 years, median RFS was longer in the combination group compared to monotherapy (HR 0.56, 95% CI: 0.31–1.2), with 18-month RFS rates of 79% versus 62%. Grade ⩾3 TRAEs occurred in 25% of patients in the combination arm and 18% in the monotherapy arm.
For HCC, personalized therapeutic vaccines have also been tested in the clinic with encouraging results. A phase I/II trial assessed the safety and immunogenicity of a DNA plasmid personalized vaccine (GNOS-PV02), encoding up to 40 neoantigens in combination with pembrolizumab for patients with MKI-treated advanced HCC. 123 Thirty-six patients were enrolled, with no dose-limiting toxicities or grade ⩾3 TRAEs reported. Injection site reactions were the most common AEs, affecting 86.4% of patients. Preliminary efficacy data indicated an ORR of 30.6%, with three patients (8.3%) achieving a CR. Immunogenicity analyses revealed neoantigen-specific T-cell responses and expansion of T-cell clones within tumours following vaccination, confirming reactivity against the vaccine-encoded neoantigens. 123 Given these initial findings, personalized cancer vaccines may represent a promising strategy to augment the anti-tumour immune response in conjunction with ICIs.
Future perspectives
Managing patients with HCC is challenging because it requires addressing both tumour burden and underlying cirrhosis simultaneously. Initial concerns regarding immunotherapy-induced hepatitis have been largely overcome through extensive experience in managing imAEs. 124 Consequently, expanding the indication of immunotherapy to previously deemed unsuitable populations is a logical progression, with early trials showing promising outcomes.
The development of biomarkers for HCC has been slow, which is hampered by the lack of biopsy as the majority of HCC could be diagnosed radiologically. Concerns about bleeding risks associated with liver biopsy in cirrhotic patients persist 125 ; however, contemporary studies indicate that the bleeding risk is generally below 1%.125,126 In this context, the latest ESMO guideline for HCC recommends the widespread use of biopsy for accurate diagnosis and research purposes. 127 The availability of biopsy will be vital, as it provides the opportunity to sequence the tumour DNA and RNA, to identify immuno-subtypes, which can help pinpoint potential biomarkers predictive of response and survival to immunotherapy. Additionally, while various aetiologies of HCC have shown consistent benefits from ICIs, the magnitude of benefit apparently differs, with greater benefit seen in patients with hepatitis B viral infection and less benefit in patients with non-viral aetiology. 128 This is particularly relevant given the rising incidence of HCC linked to non-viral aetiology such as metabolic syndromes or alcoholism.129,130
While ICIs have remarkably improved survival for patients with advanced HCC, only 20%–30% of patients will respond, and the majority will eventually develop resistance, highlighting a significant clinical unmet need. With wider experiences with ICIs, it has become clear that strategies that aim to restore the body’s endogenous immune responses may be insufficient to clear all cancer cells in the majority of patients. By contrast, the more personalized approach such as with the use of CAR-T therapy, synthetically rebuilds the immunity that targets TSA, has shown great promise. Innovative designs of CAR-T therapy such as the ‘multi-antigen logic gates’ system and armouring CAR-T with the ability to neutralize the immunosuppressive effects exerted by the TME, may enhance CAR-T efficacy and limit off-target toxicity. Other novel immunotherapeutic strategies such as BiAb and therapeutic cancer vaccines are being actively explored and early-phase clinical trials are underway.
Finally, as multiple immunotherapies emerge, determining the optimal treatment sequence becomes a relevant clinical question. A global research consortium is needed to collate and maintain a high-quality real-world database to provide insights that randomized clinical trials may not address.