
Abstract
Background:
Circulating tumor DNA (ctDNA) has emerged as a biomarker that can define the risk of recurrence after curative-intent surgery for patients with colorectal cancer (CRC). However, beyond the predictive power of postoperative ctDNA detection, the efficacy and potential limitations of ctDNA detection urgently need to be fully elucidated in a large cohort of CRC.
Objectives:
To define potentially cured CRC patients through ctDNA monitoring following surgery.
Design:
A prospective, multicenter, observational study.
Methods:
We enrolled 309 patients with stages I–IV CRC who underwent definitive surgery. Tumor tissues were sequenced by a custom-designed next-generation sequencing panel to identify somatic mutations. Plasma was analyzed using a ctDNA-based molecular residual disease (MRD) assay which integrated tumor-genotype-informed and tumor-genotype-naïve ctDNA analysis. The turnaround time of the assay was 10–14 days.
Results:
Postoperative ctDNA was detected in 5.4%, 13.8%, 15%, and 30% of patients with stage I, II, III, and IV disease, respectively, and in 17.5% of all longitudinal samples. Patients with positive postsurgery MRD had a higher recurrence rate than those with negative postsurgery MRD [hazard ratio (HR), 13.17; p < 0.0001], producing a sensitivity of 64.6%, a specificity of 94.8%, a positive predictive value (PPV) of 75.6%, and a negative predictive value (NPV) of 91.5%. Furthermore, patients with positive longitudinal MRD also had a significantly higher recurrence rate (HR, 14.44; p < 0.0001), with increased sensitivity (75.0%), specificity (94.9%), PPV (79.6%), and NPV (93.4%). Subgroup analyses revealed that adjuvant therapy did not confer superior survival for patients with undetectable or detectable MRD. In addition, MRD detection was less effective in identifying lung-only and peritoneal metastases.
Conclusion:
Postoperative ctDNA status is a strong predictor of recurrence independent of stage and microsatellite instability status. Longitudinal undetectable MRD could be used to define the potentially cured population in CRC patients undergoing curative-intent surgery.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide. 1 Surgical resection is currently the standard treatment for most nonmetastatic diseases and a subset of metastatic patients. With a curative-intent surgical procedure, more than 80% of patients with stage II colon cancer and nearly 50% of patients with stage III colon cancer are cured by surgery alone.2–6
To increase the cure rate of CRC after radical resection, continuous efforts have been made to improve the adjuvant treatment model in recent decades.7–9 To eliminate potential minimal/molecular residual disease (MRD), guidelines recommend identifying patients with high risk following curative-intent therapy who may benefit from additional adjuvant therapy by several assays. 10 In recent years, circulating tumor DNA (ctDNA) has emerged as a promising noninvasive biomarker for MRD detection following curative-intent treatment in CRC and other cancer types.11–13 Detection of persistent ctDNA after surgery or adjuvant treatment was associated with a high risk of recurrence in CRC.11,14–26 Much more attention is currently focused on detectable MRD to select patients who are at high risk of cancer persistence or disease progression.
Recently, the DYNAMIC-II study (ACTRN12615000381583) which recruited 455 stage II colon cancer patients showed that chemotherapy de-escalation or escalation according to postoperative ctDNA status could reduce adjuvant chemotherapy use from 28% to 15% in these patients, and recurrence-free survival was not compromised. 9 This proof-of-concept study confirmed the very low risk of recurrence in stage II colon cancer patients who were ctDNA negative at week 4 and week 7 after surgery, even if they were untreated after surgery.
Furthermore, it was reported that longitudinal undetectable MRD could define potentially cured non-small-cell lung cancer patients by surgery. 12 So we used the same strategy to observe the long-term clinical outcomes of CRC patients with postsurgery or longitudinal undetectable MRD in this prospective, multicenter real-world study. We recruited a heterogeneous cohort of patients with stage I–IV colorectal cancer, with the following key questions: First, whether this MRD assay could stratify CRC patients for risk of recurrence? Second, what is the negative and positive predictive value (PPV) of ctDNA in predicting disease recurrence? Third, what might affect the accuracy of ctDNA prediction of recurrence?
Methods
Participant enrollment
This prospective multicenter real-world study recruited patients with stage I–IV CRC from February 2017 to February 2021 at Sun Yat-Sen University Cancer Center, the First Affiliated Hospital of Shantou University Medical College, and the First Affiliated Hospital of Guangxi Medical University in China. Eligible patients were aged ⩾18 years and had no malignant tumor history within the past 5 years. The exclusion criteria are as follows: (1) R1 and R2 resection during operation and (2) unqualified blood samples caused by hemolysis, coagulation, insufficient specimen volume, and other factors. Tumor tissue was collected at surgery and blood samples were collected at 3–14 days after surgery or before the initiation of adjuvant chemotherapy, and every 3–6 months according to clinicians’ and patients’ decisions. Longitudinal time point was defined as serial postoperative time points until disease recurrence. All patients were treated and followed up according to the Chinese Society of Clinical Oncology Guideline. 27 Clinicopathological data, postoperative follow-up, and surveillance information were collected and analyzed.
Targeted capture sequencing
Twenty milliliters of peripheral blood in two 10-mL Streck tubes was collected at each time point. Within 3 days, the sample was separated by centrifugation at 1600×g for 10 min, and the supernatant was transferred to microcentrifuge tubes and centrifuged again at 16,000×g for 10 min to remove cell debris. Genomic DNA from tissues and peripheral blood cells and cfDNA from plasma were extracted using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), respectively. Sequencing libraries were constructed using the KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA) as per the manufacturer’s instruction. Unique identifiers (UIDs) were tagged on each double-stranded DNA to distinguish authentic somatic mutations from artifacts, improving the ability to precisely track individual plasma molecules. Barcoded libraries were hybridized to a custom-designed 1021 panel for tissue samples and a 338 panel for plasma samples, along with their paired genomic germline DNA, as previously reported. 12 The indexed libraries were sequenced with a 100-bp paired-end configuration on a DNBSEQ-T7RS sequencer (MGI Tech, Shenzhen, China) or Gene+Seq-2000 sequencing system (GenePlus-Suzhou, Suzhou, China).
Genomic data analysis
Genomic data analysis was performed as previously reported.12,28 Sequencing data were analyzed using default parameters. Adaptor sequences and low-quality reads were removed. The clean reads were aligned to the reference human genome (hg19) with Burrows-Wheeler Aligner (version 0.7.12-r1039; http://bio-bwa.sourceforge.net/). Duplicated reads were marked and removed using the MarkDuplicates tool in Picard (version 4.0.4.0; Broad Institute, Cambridge, MA, USA) for tumor and germline genomic DNA. For cfDNA, duplicated reads were identified by UID and the position of template fragments to eliminate errors introduced by polymerase chain reaction (PCR) or sequencing using realSeq (v3.1.0 Geneplus-Beijing, in-house, Beijing, China). Local realignment around single-nucleotide variants (SNVs) and indels, as well as quality control assessment, was performed using GATK (version 3.4.46; Broad Institute, Cambridge, MA, USA). Tumor somatic SNVs and small insertions and deletions were profiled using realDcaller (v1.8.1 Geneplus-Beijing, in-house, Beijing, China) and TNscope (v3.8.0 Sentieon Inc., San Jose, CA, USA). CNVKit was used to detect copy number alterations. The structural variations were analyzed using the self-developed algorithm NCsv (version 0.2.3 Geneplus-Beijing, in-house, Beijing, China). Detailed variant calling and filter strategies were reported previously. 12 The final candidate variants were all manually verified using Integrative Genomics Viewer.28,29 Targeted capture sequencing required a minimal mean effective depth of coverage of 500× in tumor tissues and 3000× in plasma samples.
CtDNA-MRD detection
A set of ~500 healthy individual plasma samples was sequenced to construct a background VAF distribution model for each target SNV. After sequencing errors were polished by UID, SNV calling was performed using a custom bioinformatics pipeline optimized for ultra-low-frequency mutation calling as previously reported. 12 SNV and indel calling was carried out by both realDcaller and TNscope (Sentieon Inc., San Jose, CA, USA) to improve the detection of long indels. After annotation, variants met any of the following criteria were filtered out: (1) the variants present in matched genomic DNA; (2) the single-nucleotide polymorphisms at >1% population allele frequency in ExAc or 1000 Genomes Project; and (3) with positional depth lower than 300×.
Based on whether the allele was identified in matched tumor tissue, two different methods were used to call plasma cfDNA variants. For tissue-derived variants in plasma, the variants showed a statistically significant difference in background errors, which was considered reliable. Meanwhile, tumor-specific driver mutations required at least two good support reads, and for other non-recurrent variants, a minimum supporting read of four. For cfDNA variants not occurring in matched tumor tissue, if the following stringent conditions were met, they were considered to be true somatic mutations: (1) for hotspot mutations, ⩾4 high-quality support reads, or for non-hotspots, at least ⩾8 support reads and (2) clonal hematopoiesis were filtered through deep sequencing of paired white blood. 12 A plasma sample with at least one variant detected was defined as ctDNA positive.
Statistical analysis
Disease-free survival (DFS) was measured from the day of definitive surgery to the first radiographic recurrence or death. Analysis of the PPV and NPV was completed for patients with at least 1 year of follow-up since the first detectable or undetectable MRD. The Kaplan–Meier method was used to describe the survival outcomes. A log-rank test was used for hazard ratios, and all p-values were based on two-sided testing with statistically significant differences at p ⩽ 0.05. Pearson’s χ2 test or Fisher’s exact test was employed to compare the difference in categorical clinicopathologic characteristics. One-to-one propensity score matching (PSM) was applied to reduce selection bias in patients with undetectable MRD who received adjuvant therapy. Multivariate Cox proportional hazards regression analysis was performed to evaluate the independent prognostic factors of DFS. Statistical analysis was performed using R software (version 4.2.0; R Foundation for Statistical Computing, Vienna, Austria) or GraphPad PRISM 9.0 (GraphPad Software, San Diego, CA, USA).
Results
Patient characteristics and tissue mutation identification
Patient enrollment and study overview are presented in Figure 1(a). A total of 330 patients diagnosed with clinical stage I–IV CRC were recruited at the study entry, and 309 patients with curative-intent surgery were retained for analysis. Patient characteristics (12.3% stage I, 43.0% stage II, 38.2% stage III, and 6.5% stage IV) were shown (Supplemental Table 1). There were 221 colon cancer and 88 rectal cancer patients and a total of 191 (61.8%) patients received adjuvant therapy, which is not different between colon cancer and rectal cancer cohort. The median follow-up period was 19.5 months (IQR, 12.1–30.0 months) with recurrence occurring in 16.8% (52/309, 2.6% in stage I, 14.3% in stage II, 20.3% in stage III, and 40.0% in stage IV) of patients.
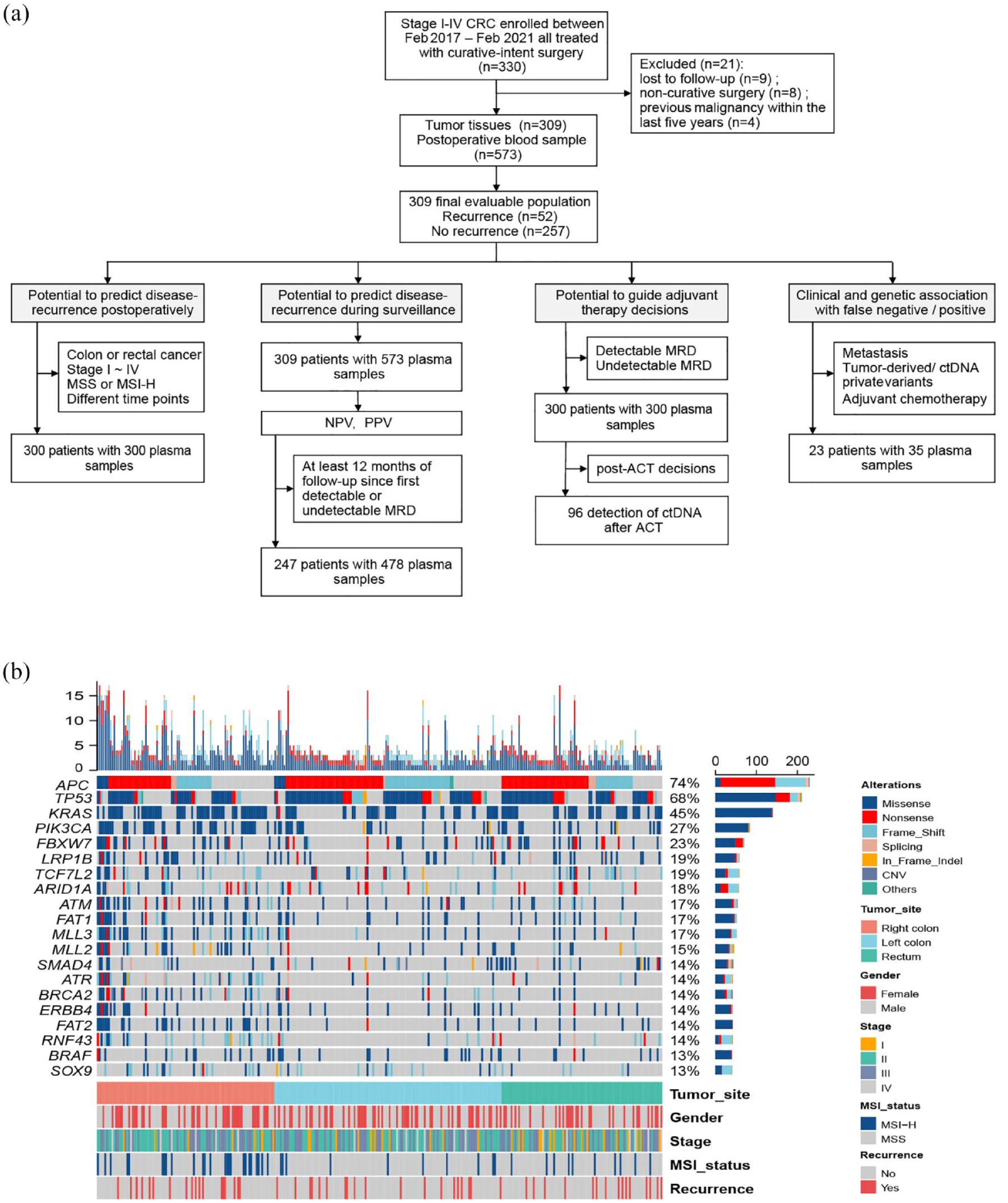
Study design and mutation landscape: (a) Flow diagram of patient inclusion in subanalyses, with the number of patients and plasma samples annotated. (b) Mutation landscape of tumor tissues and clinical characteristics of each patient. The number of somatic mutations of each patient and the mutation frequency of each gene are shown at the top and right, respectively. The bottom heatmaps indicate key patient clinical characteristics.
We initially subjected the resected primary tumor tissue to targeted massively parallel sequencing using the previously described 1021-gene assay12,28 and identified at least 1 somatic mutation in 307 of the 309 (99.4%) primary tumor tissue samples analyzed. Overall, 9722 variants were identified in tumor tissues, including 8287 SNVs, 1261 insertions/deletions (indels), 3 structural variations, and 171 somatic copy number variants [CNVs; Figure 1(b)].
Postoperative ctDNA status and recurrence in patients
For the postoperative ctDNA analysis, a single plasma specimen was collected after surgery at the median of 8 days (IQR, 6–13 days). As 9 patients did not get plasma before the start of adjuvant therapy, 300 patients were included for the postoperative ctDNA analysis. Post-surgery plasma ctDNA levels were quantified using a previously validated ctDNA analysis pipeline, tracking tumor-derived variants and non-tumor-derived variants in plasma. 12 Of the 300 patients, 43 (14.3%) patients had detectable ctDNA after surgery, including 5.4% (2/37) of patients with stage I, 13.8% (18/130) with stage II, 15.0% (17/113) with stage III, and 30.0% (6/20) with stage IV disease. The recurrence rate was 72.1% for the ctDNA-positive patients (31/43), in contrast to 6.6% (17/257) for the negative patients. DFS for these ctDNA-positive patients was significantly shorter than those ctDNA-negative patients [hazard ratio (HR), 13.17; 95% confidence interval (CI), 5.54 to 31.29; p < 0.0001; Figure 2(a)]. No correlation was observed between the presence of detectable ctDNA and clinicopathological parameters including localization, stage, and microsatellite instability (MSI) status (Supplemental Figure 1A-C). Regarding gene mutations in the primary tumor, detectable ctDNA was more frequent in patients with ZFP36L2, or GAB2 mutations; conversely, an inverse correlation between CTNNB1 mutations and postoperative ctDNA was identified (Supplemental Figure 1D).

Postoperative ctDNA status and recurrence risk. (a) Kaplan–Meier analysis of DFS stratified by ctDNA detection after surgery: detectable (n = 43) versus undetectable (n = 257). (b) Kaplan–Meier analysis of DFS stratified by both ctDNA detection and colon/rectal cancer (colon cancer, n = 217; rectal cancer, n = 83). (c) Kaplan–Meier analysis of DFS stratified by both ctDNA detection and clinical stages. (d) Kaplan–Meier analysis of DFS stratified by MSI status (MSS, n = 256; MSI-H, n = 44). (e) Kaplan–Meier plot of DFS stratified by both ctDNA detection and MSI status. (f) Concentration of postoperative ctDNA in patients with different groups of blood sample timing. p Value from Wilcoxon test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (g) Detection rate of postoperative ctDNA in patients with different groups of blood sample timing (⩽1 week, n = 142; 1–2 weeks, n = 93; >2 weeks, n = 65). (h) Kaplan–Meier analysis of DFS stratified by both ctDNA detection and sample timings.
Comparable results were observed between colon cancer and rectal cancer [Figure 2(b)], as well as among different stages of CRCs [Figure 2(c)]. In addition, as expected, MSS patients in our cohort had worse DFS compared to MSI-H patients [HR, 4.95; 95% CI, 2.36–10.39, p = 0.013; Figure 2(d)], and postoperative ctDNA could further stratified MSS or MSI-H patients into cohorts with significantly different risk of recurrence [HR, 12.89; 95% CI, 5.41–30.69 for MSS; p < 0.0001; HR, 10.49; 95% CI, 0.077–1431 for MSI-H; p = 0.038; Figure 2(e)]. All the data above indicated that postoperative ctDNA status is a strong predictor of recurrence independent of stage and MSI status.
We further sought to investigate whether the timing of ctDNA testing after resection affected the ctDNA detectability. High levels of wild-type cfDNA induced by surgical trauma could dilute the ctDNA, potentially below the detection threshold. In fact, we observed a significant decrease in cfDNA concentration 2 weeks after surgery, as shown in Figure 2(f). And we noticed numerical but not significant higher ctDNA detection rates in the samples collected >1 week after surgery, than in the samples collected within the first 1 week of resection [p = 0.7333; Figure 2(g)]. Interestingly, when samples collected from different time points after surgery were analyzed, a higher risk of recurrence was observed in the later sample timing [HR, 8.60; 95% CI, 2.19–33.78 for ⩽ 1 week; p < 0.0001; HR, 14.01; 95% CI, 3.25–60.38 for 1–2 weeks; p < 0.0001; HR, 31.23; 95% CI, 6.06–161 for >2 weeks; p < 0.0001; Figure 2(h)].
Longitudinal ctDNA, NPV/PPV
Next, we explored whether longitudinal ctDNA analysis improved recurrence prediction compared with a single ctDNA analysis. A total of 573 postoperative blood samples were collected, with an average of 1.9 times per patient (range, 1–7). Patients were defined as longitudinally ctDNA positive if they were ctDNA positive at any timepoint. Overall, 54 of 309 (17.5%) patients had detectable ctDNA during the follow-up. Compared with patients who had undetectable ctDNA during the follow-up, these patients had significantly shorter DFS [median DFS 20.5 months versus unreached; HR, 14.44; 95% CI, 7.03–29.66, p < 0.0001, Figure 3(a)]. Univariate Cox regression analysis of clinical and genetic factors showed that tumor stage, MSI status, adjuvant therapy, MLL3 mutations, ATR mutations, and longitudinal ctDNA status were all significantly associated with DFS, whereas the later multivariate analysis revealed that ctDNA status was the most significant prognostic factor (Supplemental Table 2).

MRD monitoring after surgery. (a) Kaplan–Meier analysis of DFS stratified by longitudinal ctDNA detection: detectable (n = 54) versus undetectable (n = 255). (b) Comparison of time to relapse by ctDNA and CT. (c) The NPV and PPV of undetectable and detectable MRD at postsurgery and longitudinal time points, respectively. (d) NPV and PPV of undetectable and detectable MRD at longitudinal time points across different stages.
For 39 patients, ctDNA was detected in samples collected during surveillance before relapse, and in those cases, the median lead time between ctDNA detection and imaging-confirmed recurrence was 10.1 months [Figure 3(b)].
To explore the long-term value of postsurgery and longitudinal ctDNA in predicting disease, 60 and 62 patients were excluded from further analysis, respectively, because of insufficient follow-up (less than a year since the first ctDNA negative or positive, Figure 1). In the postsurgery time point analysis, 199 patients (82.9%) had undetectable ctDNA. Most of them (n = 182) remained disease free, with an NPV of 91.5%. Conversely, the PPV was 75.6% (31/41), with a sensitivity of 64.6% (31/48), and a specificity of 94.8% (182/192; Supplemental Figure 2). Moreover, when integrating longitudinal time points, the NPV, PPV, sensitivity, and specificity were further elevated to 93.4%, 79.6%, 75.0%, and 94.9%, respectively [Figure 3(c) and Supplemental Figure 2]. The NPV of longitudinal ctDNA detection retained remarkably high levels at different stages: 100% in stage I; 96.1% in stage II; 90.0% in stage III; and 80.0% in stage IV (Figure 3D).
Predictive value for adjuvant therapy decisions
Emerging data demonstrated that apart from a prognostic factor, ctDNA-based MRD may also be a predictive biomarker for adjuvant therapy. Considering the high NPV in our cohort, we investigated the role of adjuvant therapy in undetectable MRD patients and found that adjuvant therapy could not improve DFS [Figure 4(a)]. It is worth noting that patients with adjuvant therapy tended to be at a later stage (Supplemental Table 3); thus, the PSM was used to balance potential baseline variables between the two groups, and a similar result was further confirmed [Figure 4(b); Supplemental Table 3]. We further analyzed 43 patients with detectable MRD at pre-adjuvant timepoints with or without adjuvant therapy. Confusingly, adjuvant therapy was also not found to confer a superior survival for patients with detectable MRD [HR, 1.20; 95% CI, 0.49–2.96, p = 0.70, Figure 4(c)]. However, this observation was preliminary due to the small sample size (Supplemental Table 4). In addition, both groups showed a numerical but not significant higher rate of recurrence: 74.3% (26/35) for the patients with adjuvant therapy, and 62.5% (5/8) for those without adjuvant therapy [p = 0.6649; Figure 4(d)]. We speculated that the current adjuvant chemotherapy (ACT) strategies were insufficient for this high-risk group. Then, we further analyzed the clearance of ctDNA for 14 patients with detectable MRD who received adjuvant therapy and had plasma after ACT. Only 5 (35.7%) patients presented a complete clearance of ctDNA at the end of ACT and had no evidence of relapse in further follow-up. However, all the nine patients, who did not clear their ctDNA, relapsed [Figure 4(e)]. When considering all ACT-treated patients with a post-ACT plasma available (n = 96), significantly shorter DFS was observed in ctDNA-positive patients after completing ACT than in ctDNA-negative patients [HR, 6.34; 95% CI, 2.36–17.03, p < 0.0001, Figure 4(f)].

MRD as a predictive biomarker for adjuvant therapy decisions. (a) Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with undetectable MRD at preadjuvant and postoperative time points: with adjuvant therapy (n = 147) versus without (n = 110). (b) After PSM, Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with undetectable MRD at preadjuvant and postoperative time points: with adjuvant therapy (n = 69) versus without (n = 69). (c) Kaplan–Meier analysis of DFS stratified by adjuvant therapy for patients with detectable MRD at preadjuvant and postoperative time points: with adjuvant therapy (n = 35) versus without (n = 8). (d) Recurrence rate of patients with detectable MRD at preadjuvant and postoperative time points: with adjuvant therapy (n = 35) versus without (n = 8). (e) Recurrence status of patients whose ctDNA was cleared (n = 5) or not cleared (n = 9) after adjuvant chemotherapy. (f) Kaplan–Meier analysis of DFS of the 96 patients who had plasma samples drawn after ACT, stratified by ctDNA status at the first sampling point post-ACT.
Clinical and genetic association with false negative/positive
To investigate the causes of false positives and false negatives, we analyzed the concordance between the clinical courses and the ctDNA status of 23 patients [Figure 5(a)].

Subanalysis of patients with false-positive and false-negative ctDNA analysis. (a) Overview of treatment and blood samples analyzed for ctDNA in 23 patients with false positive (n = 10) or false negative (n = 13). (b) Number of patients whose ctDNA contained the tumor-derived mutations. (c) Prevalence of genes with higher mutation frequency in primary CRC tumors with different groups of metastatic sites. (d) The distribution of mutant pathways in primary tumors with different groups of metastatic sites. (b–d) The asterisk above the bar indicates a significant difference between groups with p value less than 0.05 determined by the chi-square test.
Ten of the 23 patients were disease free despite detectable MRD after surgery (6 stage II and 4 stage III). Of these 10 patients, 4 patients were treated with capecitabine for a medium of 3 months (3 in stage II and 1 in stage III), and the other 4 patients were treated with oxaliplatin-based doublets (1 in stage II and 3 in stage III). Notably, four recurrence-free patients with available serial plasma samples showed ctDNA clearance after ACT. Four of the patients who received ACT had no other MRD testing besides the postsurgery MRD analysis. Thus, there might be some patients who benefited from ACT and counted for the false-positive MRD result of the postsurgery analysis.
We then analyzed the 13 patients who relapsed with undetectable MRD (3 stage II, 8 stage III, and 2 stage IV) with a medium DFS of 11.6 months. Undetectable MRD may be associated with the site of recurrence because six of them had lung metastases and four patients had peritoneal metastases. This encouraged us to investigate whether different metastatic sites had different MRD patterns. So we compared the MRD mutation profiling of patients with disease recurrence (n = 52). Remarkably, 11 of the 15 patients with lung metastasis had no detectable tumor-derived mutations (5 with ctDNA-private mutations, 6 with undetectable mutations), and 6 of the 15 patients with lung metastasis had non-tumor-derived mutations, while 10 of the 13 patients with liver metastasis had tumor-derived mutations [Figure 5(b)]. We further compared genomic profiles of the primary tumors, and less APC mutations in the peritoneal metastasis group were detected (Figure 5C). In line with this observation, we detected increased Wingless/int1 (WNT) signaling activity in primary tumors that would eventually recur as distant metastatic disease [e.g. liver and/or lung metastases; Figure 5(d)]. Moreover, a significant enrichment of the TGF-β signaling pathway was present in the liver metastasis group, compared to the lung and peritoneal metastasis groups [Figure 5(d)].
Discussion
The results from 309 patients were consistent with previous studies that positive ctDNA status could reflect the existence of MRD and thus the significantly higher risk of disease recurrence.11,17,18,22,23,30 It is worth noting that about 75–100% of patients with longitudinal undetectable MRD remained disease free in these studies, indicating that they may have been cured. Identifying those patients to avoid potential overtreatment is also a significant clinical concern besides identifying patients who were at high risk of recurrence. Therefore, we highlighted the NPV of MRD detection in this study.
Patients with undetectable MRD in our study could maintain a high disease-free rate across I–IV stages (91.5% for postsurgery analysis and 93.4% for the longitudinal analysis). For patients of stage II, up to 40% of patients undergo adjuvant therapy in routine clinical care 31 ; however, only an absolute risk reduction of 3–5% in this population was reached. 18 So, the high NPV of stage II (96.1%) may benefit patients by sparing them from unnecessary drug toxicity, economic burden, and even radiological exposure. Moreover, our NPV was comparable to the DYNAMIC-II study, in which recurrence or death occurred in 15 of 246 ctDNA-negative patients (with NPV 93.9%). 9
However, the PPV in our study was exceptionally lower compared with some studies (79.6% versus 93.3–100%),11,16,24,32 and it varied across different stages. We speculated that the major reason might be the clearance of ctDNA. The definition of longitudinal positive that we used considered any instance of a positive MRD result. However, it is important to note that 40% (4/10) of patients subsequently achieved MRD negativity through treatment, resulting in no recurrence. Furthermore, when we restricted our analysis to patients with serial plasma samples collected both before and after ACT, we observed that all nine patients who did not achieve clearance of their ctDNA experienced relapse, resulting in a PPV of 100% (Figure 4(e)). The critical role of post-ACT risk stratification and management is emphasized. Insufficient follow-up time might be another potential reason. In our study, we observed that 10 patients remained disease free despite having detectable MRD at some point during longitudinal monitoring, with a median follow-up of 29.2 months (range, 15.1–42). This finding aligns with the understanding that a significant proportion of relapses occur within the first 3 years, with an additional 15% occurring between the third and fifth years. 2 Importantly, it is well established that the tumor-node-metastasis (TNM) staging system is indicative of the probability of survival across different stages. A meta-analysis has reported a 5-year DFS of 82.7% for stage II and 63.8% for stage III colon cancer. 4 These findings highlight the importance of long-term follow-up, particularly for early-stage CRC.
Only five (35.7%) patients showed persistent clearance of ctDNA with adjuvant therapy and had no evidence of relapse (Figure 4(e)). The ability of ACT to convert ctDNA positive to ctDNA negative varied greatly between different studies (16.7–67.7%),11,16–18,22,23,30 probably due to the limited sample size or different ACT regimens. However, adjuvant therapy was not found to confer a notable improvement in the DFS of patients who had detectable ctDNA postoperatively in our study. Among those who received adjuvant chemotherapy, the majority of patients received oxaliplatin-based therapy (Supplemental Table 4). Three of four patients (75%) with MSI-H tumors received adjuvant chemotherapy, two (66.7%) of whom were treated with oxaliplatin-based combination chemotherapy. Our study showed that postoperative ctDNA could further stratify MSI-H patients but chemotherapy has very limited efficacy in MSI-H tumors as reported .33,34 This high-risk group may benefit from more effective treatment options. Multiple prospective and interventional studies using different adjuvant treatments are ongoing to appraise the utility of ctDNA-guided treatment approaches in CRC. 35
Of note, a small proportion of patients who were ctDNA negative postoperatively or during the follow-up got relapse (8.5–6.6% in this study, 11.9–3.3% reported by Reinert et al. 11 ). As our data showed, the sensitivity of MRD monitoring is limited in patients with lung-only recurrence, and followed by peritoneal metastasis (Figure 5(a)). Similarly, in a study, a false-negative ctDNA assessment at the postoperative timepoint is more commonly observed in patients who subsequently experienced local relapse alone (such as peritoneal relapse) compared to those with distant relapse. 19 Several studies in metastatic CRC have shown that patients with lung and peritoneal only metastases are more likely to have undetectable ctDNA in plasma compared to patients with liver metastases,36–38 which leads to false negatives. This phenomenon could account for the evolution of tumor cells and specific routes of dissemination (e.g. via circulation or intraperitoneal seeding). Our data showed that CRC patients with lung-only metastasis had more non-tumor-derived mutations than those with liver-only metastasis. Even though a bioinformatics pipeline integrated tumor-genotype-informed and tumor-genotype-naïve ctDNA analysis, false-negative results can still occur due to the challenge of identifying these unique cfDNA variants. On the other hand, a lower mutation rate of APC was detected in the peritoneal metastasis group. Interestingly, the loss of APC has been known to constitutively activate WNT signaling and promote the stemness of CRC cells. Therefore, we speculated distinct evolutionary paths among different types of distant metastases. Consistently, a study of comparison of metastases and matching primary CRC profiles indicated that, compared to liver metastases, peritoneal lesions showed much more similarity to their parental tumor. 39 Our results reiterated the further investigation of overcoming the blood-based barriers to MRD monitoring.
Although ctDNA detection may be useful for monitoring MRD after surgery, the analytical sensitivity of the ctDNA assays is a major challenge. In this study, the detection rate of postoperative ctDNA was 30% in patients with stage IV disease, which is relatively lower than that reported in a published study (54.5%) 24 but comparable with other studies (24.5–30.4%).26,40 Several aspects must be considered. First, 50% of the enrolled IV patients had lung-only or peritoneum-only disease in our study. Second, collecting blood samples at a median of 9.5 days in this population (8 days for the overall population) might have influenced the assay sensitivity rate. False negativity caused by the elevated levels of circulating normal cfDNA may occur if we obtained plasma for surveillance too early after surgery. Recently, increased release of normal DNA was reported to be a frequent consequence of surgical procedures. 41 While the later of sample timing, the more possibility that patients who will be discharged from the hospital may be lost to follow-up or a delay in receiving treatment. To our knowledge, no optimal time for postoperative plasma ctDNA detection was explored with clinical outcomes. According to the guidelines, adjuvant therapy should be administered as soon as the patient is medically able, generally around 4 weeks after surgery and no later than 2 months.10,27 Considering the turnaround time from blood specimen collection to ctDNA result availability, which is approximately 2 weeks, we compared plasma samples within 1 week, 1–2 weeks, and >2 weeks after tumor resection and found that >2 weeks was more related to tumor recurrence. Taking into account the study by Henricksen et al., 41 MRD detection would be more practical and meaningful at 2–4 weeks from the time of surgery with a positive ctDNA result triggering the start of adjuvant therapy within the time frame recommended in guidelines.
The present study had certain limitations. First, the follow-up duration was relatively short with a median of 19.5 months. This led to only 52 cases of recurrence in our study. Second, MRD follow-up was not strictly conducted every 3–6 months after surgery. With only very limited patients having post-ACT ctDNA analysis, our data added a little to the question of whether ctDNA-positive patients would benefit more from future adjuvant regimens compared with other studies.11,17,32,42
Conclusion
We confirmed the prognostic value of ctDNA-based MRD detection in CRC patients with stage I–IV after radical resection in this prospective study. We also highlighted the value of undetectable MRD, which could be used to define the potentially cured population in CRC patients undergoing curative-intent surgery. Moreover, subgroup analysis suggested that cases with lung-only or peritoneal metastases were a major unmet challenge for MRD monitoring in CRCs.
Supplemental Material
sj-docx-1-tam-10.1177_17588359231220607 – Supplemental material for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer
Supplemental material, sj-docx-1-tam-10.1177_17588359231220607 for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer by Wenhua Fan, Zhiyuan Xia, Rongrong Chen, Dagui Lin, Fang Li, Yang Zheng, Jiongyong Luo, Yuanyuan Xiong, Pengli Yu, Wei Gao, Yuhua Gong, Feiran Zhang, Sen Zhang and Liren Li in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-jpg-2-tam-10.1177_17588359231220607 – Supplemental material for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer
Supplemental material, sj-jpg-2-tam-10.1177_17588359231220607 for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer by Wenhua Fan, Zhiyuan Xia, Rongrong Chen, Dagui Lin, Fang Li, Yang Zheng, Jiongyong Luo, Yuanyuan Xiong, Pengli Yu, Wei Gao, Yuhua Gong, Feiran Zhang, Sen Zhang and Liren Li in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-jpg-3-tam-10.1177_17588359231220607 – Supplemental material for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer
Supplemental material, sj-jpg-3-tam-10.1177_17588359231220607 for Circulating tumor DNA analysis predicts recurrence and avoids unnecessary adjuvant chemotherapy in I–IV colorectal cancer by Wenhua Fan, Zhiyuan Xia, Rongrong Chen, Dagui Lin, Fang Li, Yang Zheng, Jiongyong Luo, Yuanyuan Xiong, Pengli Yu, Wei Gao, Yuhua Gong, Feiran Zhang, Sen Zhang and Liren Li in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.