
Abstract
Background:
Pemetrexed and cisplatin is a first-line standard in non-squamous non-small-cell lung cancer without targetable mutations. It became the backbone of checkpoint-inhibitor–chemotherapy combinations. Single high doses of cisplatin pose toxicity risks and require hyperhydration, potentially prolonging outpatient application. The aim of this study was to compare efficacy, safety and tolerability of split-dose cisplatin with the standard schedule.
Methods:
Patients with metastatic non-squamous non-small-cell lung cancer were randomly assigned to up to six 21-day cycles of pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 on day 1 (arm A), or pemetrexed 500 mg/m2 (day 1) and cisplatin 40 mg/m2 (day 1 + 8, arm B), followed by pemetrexed maintenance. Primary endpoint was objective response rate. Secondary objectives were overall survival, progression-free survival, time to progression, treatment compliance, toxicity profile, and quality of life.
Results:
We enrolled 130 patients (129 evaluable). Median cycle numbers in A and B were six (1–6) and five (1–6). Dose intensities were comparable between arms. More patients in A received pemetrexed maintenance (24.2% versus 11.1%). With 16 (24.2%) in A and 19 (30.2%) patients in B achieving objective responses [odds ratio 0.74 (0.34–1.62), p = 0.55] the primary endpoint was met. Overall survival was not different between arms (median 14.4 versus 14.9 months); [HR = 1.07; (0.68–1.68), p = 0.78]. Median progression-free survival was 7.0 months in A and 6.2 months in B [HR = 1.63; (1.17–2.38); p = 0.01]. Adverse events of CTCAE grade ⩾3, particularly hematological, were more frequent in B. No difference in grade 4 and 5 infections between arms was noted. Treatment-related asthenia and nausea/vomiting of any grade were more frequent in A. Global health status, fatigue and constipation measured on day 1 of cycle 4 demonstrated superior scores in B.
Conclusion:
Pemetrexed and split-dose cisplatin is safe and effective. Advantages of split-dose cisplatin with regard to specific toxicities allow personalization of this important chemotherapy backbone.
Trial Registration:
European Clinical Trials Database (EudraCT) number 2011-001963-37.
Introduction
The doublet of cisplatin and pemetrexed represents a widely accepted standard for the treatment of advanced non-squamous non-small-cell lung cancer (NsqNSCLC) without driver mutations.1,2 It has also become the first approved chemotherapy backbone in combination with a checkpoint-inhibitor (pembrolizumab) for first-line treatment. 3 Cisplatin combinations still have a number of supporters in Europe and elsewhere, especially as cisplatin demonstrates better objective remission and by trend improved survival rates compared with the corresponding carboplatin doublets according to an individual patient-based meta-analysis. 4 Even in modern chemoimmunotherapy trials a continuing benefit regarding hazard ratios has been observed for cisplatin doublets compared with their carboplatin counterparts; the reason for this might be patient selection or rather a superior effectiveness of cisplatin in combination with pembrolizumab compared with carboplatin.3,5–8
Due to potential nephrotoxicity of cisplatin and more frequent nausea and vomiting, a standard higher-dose cisplatin is regularly administered with prolonged hyperhydration and modern antiemetic support.1,2 A split-dose cisplatin schedule can generally more easily be administered in an outpatient setting. 9
A split-dose cisplatin schedule has been reported for doublets with gemcitabine, docetaxel, etoposide, vinorelbine and paclitaxel.10–18 These administration schedules are more frequently used and are well tolerated also for outpatient administration. Split-dose cisplatin with etoposide has turned out highly effective in testicular cancer and also in adjuvant treatment of NSCLC where in both situations high efficacy has to be achieved for cure with required adequate patients’ tolerance and compliance.16,17,18,19 At the moment, for the split-dose combination with pemetrexed we do not have adequate prospective evidence. 20 Therefore, we conducted a signal-finding, randomized phase II trial to demonstrate efficacy and efficacy/toxicity ratio of such an experimental cisplatin split-dose schedule versus the standard three-weekly regimen.
Methods
Patients
All patients (pts) had to have histologically or cytologically confirmed diagnosis of NsqNSCLC, stage IVA or IV B according to the UICC version 7 (2009) and no prior systemic treatment. Patients with ECOG performance status 0 or 1, age between 18 and 75, and adequate organ functions were included. Untreated brain metastases were exclusion criteria. Patients with NsqNSCLC known to harbor EGFR-mutations were excluded. When treatment was started and prior EGFR-testing results became available, these patients with EGFR-mutations could remain on study if clinical benefit was clearly documented.
Trial oversight
The trial was registered in the European Clinical Trials Database (EudraCT) as number 2011-001963-37. Monitoring, data management and statistical analysis were performed by the Clinical Research Organization ClinAsses GmbH (Leverkusen, Germany). Manuscript writing was performed without external writing support by the authors. The clinical trial was conducted at eleven German centers associated with “Arbeitsgemeinschaft Internistische Onkologie” (AIO).
Trial design
In this phase II study 130 patients were randomized 1:1 to receive up to six cycles of either cisplatin 75 mg/m2 and pemetrexed 500 mg/m2 on day 1 every 3 weeks (arm A) or a split-dose cisplatin of 40 mg/m2 on day 1 + 8 and pemetrexed 500 mg/m2 day 1 every 3 weeks (arm B). Maintenance therapy with pemetrexed 500 mg/m² (q d 21) was foreseen in both arms until progression or termination by the patient or treating physician. Premedication with folic acid and vitamin B12 was mandatory.
In the standard treatment arm A the pre-cisplatin hydration consisted of 2 l of either 0.9% sodium chloride or dextrose 4% in one-fifth normal saline (0.18%) over a 2-h period. During the last 30 min of the pre-treatment hydration or after the hydration, 40 mg furosemide were given as diuretic treatment. Post-cisplatin hydration in arm A was performed by administering another 2 l of sodium chloride 0.9% infusion or dextrose 4% in sodium chloride 0.18% infusion over a period of 4–12 h.
The split-dose cisplatin arm B contained a less extensive pre- and post-cisplatin hydration protocol. For pre-cisplatin hydration 1 l of either 0.9% sodium chloride or dextrose 4% in one-fifth normal saline (0.18%) were administered over a duration of 1 h. During the last 30 min of the pre-treatment hydration or after the hydration, 40 mg furosemide were given for diuresis. For post-cisplatin hydration 1 l of sodium chloride 0.9% infusion or dextrose 4% in sodium chloride 0.18% infusion over a period of about 2 h were administered in arm B.
Dose reductions due to adverse events were performed according to guidelines predefined in the protocol (see supplementary data). No cross-over between arms was allowed. Less than 4 weeks prior to first study drug, radiologic tumor assessment according to RECIST-criteria had to be performed. Every two chemotherapy cycles, the tumor had to be re-assessed with the same method (CT scan or MRI scan) as at baseline. Response was determined according to RECIST 1.1. Partial or complete responses had to be confirmed with CT scans 4–6 weeks after first evaluation. The primary endpoint of this trial was objective response rate (ORR) by RECIST 1.1 determined in both arms. It should be demonstrated that there was no sign of inequality between the response rates of both arms.
Patient-reported outcomes: Quality of life
Quality of life as patient-reported outcome (PRO) was measured using the EORTC Quality of Life Questionnaire (QLQ-C30) after full consent from EORTC for its use. Patients were asked to fill out quality of life questionnaires at baseline, on day 1 of each cycle, and after the completion of therapy.
Statistical analysis
Statistical analysis included all randomly assigned patients following a minimum application of one study drug and these defined also the efficacy and safety population. This study explored the similarity of the two regimens with regards to response rates as primary outcome. The aim was not to demonstrate statistically significant differences in ORR; no formal comparison between the two arms was planned statistically. However, to demonstrate that there is no relevant statistical sign of inequality, the response rates of both treatment groups were compared in an explorative manner. Fisher’s Exact test was applied at the two-sided significance level of α = 0.05. Due to the number of patients in this randomized phase II trial and as it was not planned, a classical statistical demonstration of non-inferiority could not be performed as statistical analysis for the primary endpoint.
A log-rank test stratified to both arms of the study was used to compare in an explorative manner overall survival (OS), progression-free survival (PFS) and time to progression (TTP) of both treatment groups. Wilcoxon test was performed for comparison of continuous parameters. A two-sided p-value of 0.05 was considered significant.
Results
Patients and treatments
From 21 November 2012, until 10 December 2015, 130 patients with NsqNSCLC were registered and randomized. One patient had to be removed from study shortly following randomization due to rapid clinical progression prior to receiving any study drug because of withdrawal of consent (CONSORT-Figure 1). Thus, 129 evaluable patients were included both in study safety and efficacy population, 66 in arm A and 63 patients in arm B. Patient characteristics of all 129 evaluable patients are given in Table 1. There were minor imbalances with respect to gender, tumor grade and EGFR-mutation status. Due to the availability of targeted therapy options at that time, the study population was enriched for patients without targetable biomarkers and with KRAS mutated cancers (Table 1). A median of 6 (1–6) and 5 (1–6) treatment cycles were administered in arms A and B.
Patient characteristics.

Tumor (T), Node (N) and Metastatic (M), Stage are given according to UICC (7th) and (8th).
CRP, C-reactive protein; ECOG, Eastern Cooperative Oncology Group; LDH, Lactate dehydrogenase; UICC, Union internationale contre le cancer.

Consort diagram of the study. From 130 screened patients one patient had to be removed from the study population shortly after randomization due to a rapid progression and prior to receiving any study drug administration while at the same time withdrawing his study consent. This patient was not taken under consideration in the intent to treatment and safety population. 129 patients received the study protocols.
Primary endpoint
ORR was 24.2% (16/66) in arm A, and 30.2% (19/63) in arm B, which was not significant [odds ratio 0.74 (95% CI 0.34–1.62); p = 0.55]. Hence, the primary study endpoint was met.
Secondary endpoints
Disease control rate [DCR = sum of complete remission (CR), partial remission (PR) and stable disease (SD)] was also not different between both regimens. Confirmed disease control was achieved in 47 patients (71.2%) of A, and in 46 (73.0%) of B [hazard ratio (HR) 0.91 (95% CI 0.42–1.98); p = >0.85]. When the patient groups with tumors harboring known driver mutations (EGFR, ALK, ROS1) were excluded from statistical analysis, there was no significant difference in both parameters ORR and DCR between arms (Table 2).
Objective responses.

Complete remission (CR), partial remission (PR), stable disease (SD), progressive disease (PD) are defined by the Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. Objective response rate representing the primary endpoint is given as the sum of patients achieving a complete remission (CR) or a partial remission (PR) as defined by RECIST 1.1.-criteria (in bold). Disease control rate is given as the sum of patients achieving a complete remission or partial remission or stable disease as defined by RECIST 1.1.-criteria.
Driver mutation: A somatic mutation in the EGFR-gene or a translocation in the ALK- or ROS1-gene for which a specific targeted therapy (oral tyrosine kinase inhibitor) is available.
CI, Confidence interval.
In all patients the median OS was 14.6 months, the median PFS was 6.4 months. Based on randomization, the two study arms A and B showed the following results: median OS in A was 14.4 months versus 14.9 months in B [HR: 1.07 (95-CI: 0.68–1.68); p = 0.78, Figure 2]. If patients with driver mutations were eliminated from this analysis, the survival results were 13.8 months in A (n = 62 pts) and 14.9 in B (n = 60) [HR: 1.028; (95-CI: 0.651–1.624); p = 0.91] with no significant signal preferring any arm.

Kaplan–Meier curves of overall survival (a) and progression-free survival (b) for all patients in both study arms.
Analysis of PFS between arms showed a clinically marginal, but statistical significant difference with 7.0 months in A compared with 6.2 months in B [HR: 1.63; (95-CI: 1.12–2.38); p = 0.01]. When patients with driver mutations were eliminated from analysis, median PFS was 7.0 months in A versus 6.2 months in B [HR: 1.58; (95-CI: 1.07–2.33); p = 0.02]. The results for median TTP were 7.4 months in A and 6.2 months in B [HR 1.71, (95-CI: 1.15–2.55) p = 0.01].
Safety analysis and adverse events
Study-related adverse events (AEs) of all grades were documented for 64 pts (97%) in A and for 58 pts (92.1%) in B. The most frequent AEs (of any grade) were hematopoietic disorders (40.9% in A and 63.5% in B) and gastrointestinal disorders (89.4% in A and 85.7% in B). AEs leading to dose reduction or discontinuation of study drug treatment were more often observed in B, 21 patients (33.3%), versus 11 patients (16.7%) in arm A. The reasons for dose reduction/discontinuation were mainly due to hematological toxicities in arm B and related to gastrointestinal disorders in arm A. More hematological AEs of grade 3 and 4 were documented in B (Table 3). Also in B, more grade 3 (but no grade 4 or 5) infectious AEs occurred than in A. Arm A showed more grade 3 and also grade 4 and 5 gastrointestinal AEs than B. One gastrointestinal AE of grade 4 but no grade 5 was observed in arm B. The only treatment-related death of this clinical trial was observed in arm A following an intestinal perforation most likely related to mucositis caused by pemetrexed.
Adverse events.

Adverse events evaluated according to Common Terminology Criteria for Adverse Events (CTCAE 4.0).
In both arms no clinical relevant renal toxicity was documented (Table 3) and creatinine clearance at baseline was comparable in the study arms [112.0 ml/min (Range 56.9–227.7 ml/min) in A and 110.0 ml/min (Range 59.7–200.9 ml/min) in B]. Compared with the last documented creatinine clearance after the end of study treatment, in both arms a decrease was seen which was significant smaller in arm B. The mean of decrease was −10.9% (Range −50.8% to 58.6%) in arm A compared with −1.3% (Range −39.1% to 40.6%) in arm B (Wilcoxon test two-sided significance level of α = 0.05; p = 0.01; Supplement Table 1).
No new safety findings were observed.
Dose intensity analysis
Treatment cycle number and dose densities of cisplatin and pemetrexed were comparable in both arms (Table 4). In arm A patients received a mean of 4.5 (range 1–6) and in B a mean of 4.3 (range 1–6) cycles. Mean cumulative cisplatin dose was 328 mg/m2 (range 72.9–456.9 mg/m2) in A and 319.9 mg/m2 (range 35.3–536.3 mg/m2) in B. Patients in A received numerically more pemetrexed than in B (mean pemetrexed dose 2221 mg/m2, range: 486.3–3035.3 mg/m2) versus 2085.8 mg/m2 (range: 440.9–3070.1 mg/m2). More patients in arm A received planned pemetrexed maintenance treatment than in B [16 pts (24.2%) versus 7 pts (11.1%)]. A trend to a lower discontinuation rate of therapy could be observed in arm A [33 pts (50%) versus 38 pts (60%) in arm B)].
Dose intensity analysis.

m2, Body surface area given in m2.
Patient-reported outcomes (PROs)
Baseline QLQ-C30 questionnaires were completed by 41 (62.1%) patients in arm A, and 48 (76.2%) patients in arm B. Another 34 (51.5%, arm A) and 47 (74.6%, arm B) patients completed least one additional questionnaire between baseline and end of treatment visits. Mean values of QL2 score at baseline were comparable in both arms with 49 [±26.6 standard deviation (SD)] in arm A and 56.8 (±25.9 SD) in B.
Changes in the QLQ-C30 from baseline to start of cycle 4 (after three administered cycles) demonstrated an improvement for functioning scores of physical, emotional, cognitive and social functioning in arm B (Figure 3a). In contrast, patients in arm A reported a deterioration of scores of these items, except for emotional functioning. There was a trend toward improvement in the global health status in arm B. Regarding changes in single symptoms from baseline to start of cycle 4 there was a trend toward improvement of fatigue and constipation in arm B, while a trend for deterioration was observed in arm A (Figure 3b). In both arms there was an increase in nausea and vomiting during the course of therapy. However, that trend was less strong in arm B. Improvement of pain and dyspnea were seen in both arms at the beginning of cycle 4, but by trend more frequently in arm A. Analysis of changes from baseline to the last score of the QLQ C30 underlined a stronger trend toward improvement and less deterioration of general functions and symptoms in arm B (Figure 4a and b, supplement Table 2). In contrast to the findings to start of cycle 4 of therapy, the symptom of dyspnea was less improved and deteriorated stronger in arm A. A stronger improvement of the symptom of pain was reported in arm A.

Changes from baseline to beginning of cycle 4 of therapy in the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (QLQ-C30) are given for global functioning scores (a) and for single symptoms (b). Please Note that an increase of the column means for global functioning (a) an improvement of the homologous functioning and thus a “healthier” individual. For the single symptoms (b) a decrease of the column is associated with a lower symptom burden and an increase respectively with a higher symptom burden.
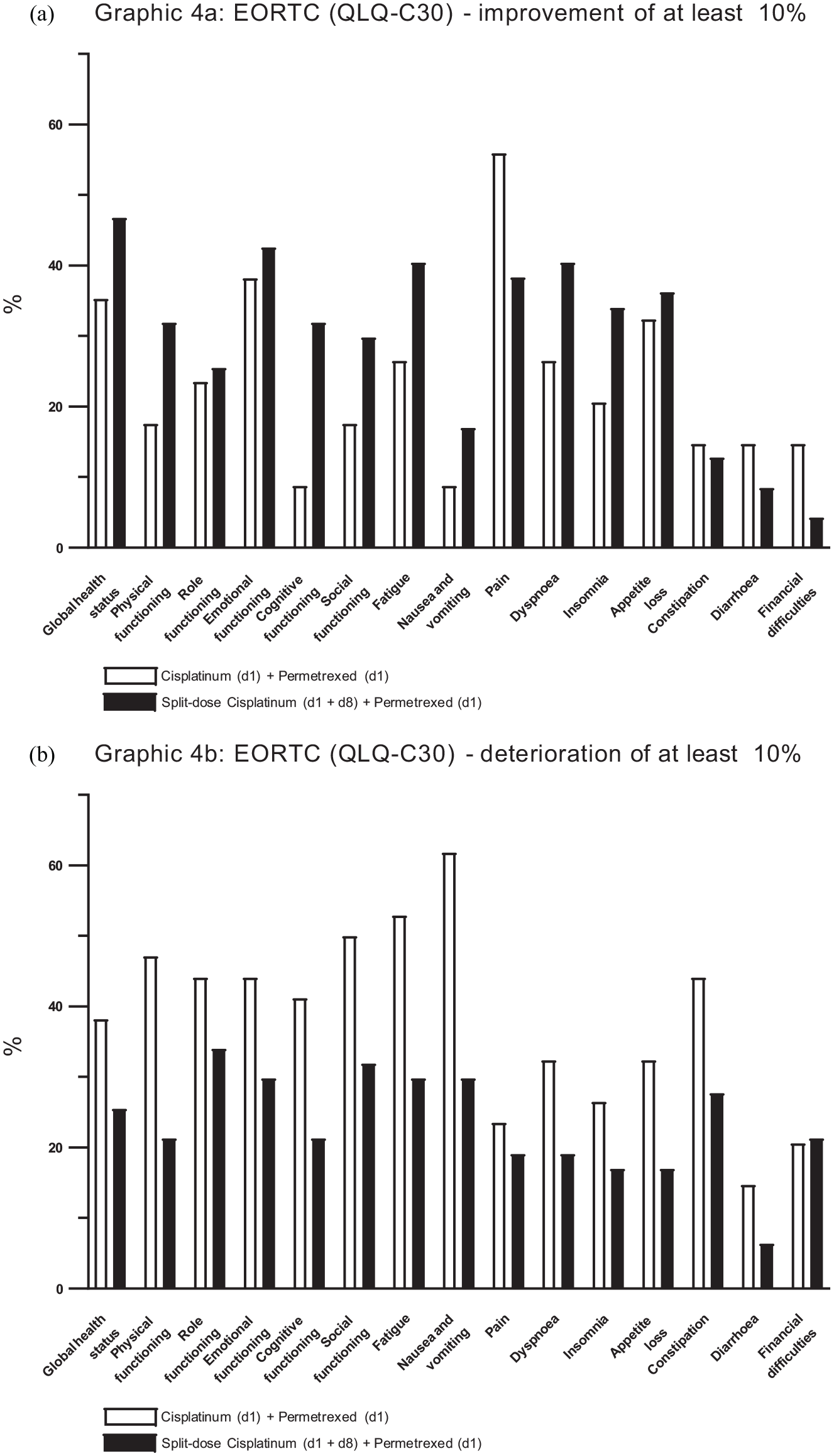
Changes from baseline to last measured score in the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire Core 30 (QLQ-C30) are given. Only patients who filled out the questionnaires were taken into the analysis. Please note that in the presented form here “Improvement” means for global functioning scores as well as for single symptoms a “healthier” individual and a lower symptom burden. A deterioration means, respectively for global functioning scores and for single symptoms, a less healthy individual and higher symptom burden. Percentages of patients who received an improvement of more than 10% (a) or a deterioration of more than 10% (b) are given. White bars represent study-arm A (cisplatin standard dose) and black bars represent arm B (cisplatin split-dose). An analysis regarding changes of more than 10% was chosen as an analysis of 10 scores or respectively 10% can considered to be the minimal important difference indicating a clinical meaningful difference. 21
Discussion
Cisplatin and pemetrexed is a well-established standard for advanced NsqNSCLC from the pre-immunotherapy era.1,2,22 It became also the first approved chemotherapy combination to be administered together with checkpoint-inhibitor (pembrolizumab) and it represents today one of the current standards in first-line therapy for patients with NsqNSCLC.1–3 Divided cisplatin doses combined with other agents have long been accepted as a valid strategy for treatment of NSCLC.10–15,18 For chemoradiotherapy of high-risk head and neck squamous carcinoma, a cisplatin split-dose regimen is better tolerated and non-inferior to the 3-weekly schedule. 23 We hypothesized that in advanced NsqNSCLC this strategy could also give some benefits and that a d1 and d 8 administration of split-cisplatin doses would improve tolerance and reduce toxicity of the cisplatin and pemetrexed doublet, this being the basis for a randomized phase II study looking at these two application schedules.
In our signal-finding randomized phase II trial the experimental d 1 and d 8 cisplatinum schedule showed an ORR of 30.2% compared with 24.2% in the standard schedule. To compare these response rates in an explorative manner, the test of inequality showed a significance level of >0.05 (odds ratio 0.74, p = 0.55) and, therefore, the equality of the response rates in both study arms cannot be ruled out and the primary endpoint of this study was met.
The median OS was 14.9 months (14.9 months in patients without driver mutations) in B and 14.4 months (13.8 months in patients without driver mutations) in A (log-rank OS: HR 1.07, p = 0.78). In summary, considering ORR and OS being the major outcome parameters, no relevant signal favored any of the two administration schedules.
Interestingly, we could observe significant differences in secondary time-to-event endpoints PFS and TTP. Median PFS of arm A was 7.0 months versus 6.2 months in arm B (HR 1.63, p-value = 0.01) and TTP was 7.4 months in arm A versus 6.2 months in arm B (HR 1.71, p-value = 0.01). This observed difference, however, cannot clearly be considered clinically meaningful. It could possibly be explained by small imbalances in the study arms, that is, the fraction of high-grade, undifferentiated lung tumors (G3 versus G2) was different between arm A versus arm B: 17/21 versus 24/16. We analyzed what could further contribute to differences in PFS and TTP and found the number of patients who received maintenance pemetrexed to be different between the two arms (Table 4). While there was no significant difference between induction treatment dose intensity for cisplatin and pemetrexed (Table 4), there were seven patients in B and 16 in A who received pemetrexed maintenance, demonstrating a clear trend for a higher usage of pemetrexed maintenance in arm A (X 2 -test, p = 0.14). Due to the slightly higher number of hematologic toxicities, fewer patients in the split-dose arm probably received a pemetrexed maintenance, which could partly explain the shorter median PFS in B, especially as pemetrexed maintenance is known, from the pivotal randomized trials, to extend the PFS. 24 The larger number of patients in arm B who underwent discontinuation or dose reduction due to an AE (33.3% versus 16.7%)—mostly hematological toxicities—might also have contributed to the shorter median PFS and TTP in arm B, but did not influence the ORR here as primary endpoint.
Patients with tumors harboring driver mutations were by trend slightly more frequent in arm A, which also might to some parts have influenced the outcome differences regarding PFS and TTP.
No new safety results were reported for either application schedule in this trial. There was by trend a higher rate of treatment discontinuation in the split-dose arm, which is most likely explained by hematologic side effects being more often reported in B than in A. However, gastrointestinal toxicities ⩾ grade 3 and nausea and vomiting have been more often reported in A than in B. When we looked at all the toxicity findings of both arms there were only minor clinical differences (Table 3). Although there seemed to be slightly more hematological toxicity in B, the number of infections and treatment-related deaths was not clearly favoring A.
In both study arms no relevant renal toxicity was documented (Table 3). However there was a decrease in the creatinine clearance after the end of study therapy in arm A and B which was significant lower in the split-dose arm B (Supplement Table 1).
Astonishingly, when looking at life quality analyses, there were several items clearly favoring the experimental schedule: global health status, physical functioning, emotional functioning, cognitive functioning and social functioning (Figure 3a). The standard arm was only superior in items pain and dyspnea (Figure 3b). The PROs therefore demonstrated a better improvement and less deterioration of quality of life in B. Considering the lower rate of gastrointestinal toxicity and of nausea and vomiting as side effects with a strong deterioration of subjective well-being and more favorable PROs, arm B can be seen in some regards better tolerated for patients in clinical practice. Besides the more favorable toxicity profile of arm B, the less extensive pre- and post-cisplatin hydration protocol in arm B made the cisplatin split-dose protocol somehow easier to administer.
When comparing our results with the literature for cisplatin and pemetrexed, Kawano et al. 25 reported a higher response rate of 44.0% (30–58%) in a single-arm phase II study in 50 Japanese patients. Nevertheless, this was a small and selected phase II population and the proportion of tumors harboring driver mutations was even higher in that trial compared with our trial, based on the increased frequency in Asia. Koba et al. 20 reported in another single-arm phase II study (53 patients; primary endpoint 1-year survival rate) for a pemetrexed (500 mg/m2 d1) and cisplatin split-dose regimen (40 mg/m2 d1 + d8) in Japanese patients with NsqNSCLC a response rate of 37.7%, a median PFS of 5.3 month and a median OS of 18.6 month. Gandhi et al. 3 reported in a large, world-wide randomized phase III study a response rate of 18.9% (13.8–25.0%) for cisplatin and pemetrexed administered at day 1 in more than 206 patients in the standard arm. Our 129 patients lie somewhere between these three results and also intermediate in the patient number.
The median OS and PFS reported here correspond to the results in other large phase II and phase III clinical trials. Scagliotti et al. 22 reported a median OS of 12.6 months and a PFS of 4.8 months. Kawano et al. 25 and Asami et al. 26 reported in their single-arm phase II studies in 50 and 35 Japanese patients a median OS of 22.0 and 15.5 months and a PFS of 4.3 and 6.7 months, respectively. More recently, Gandhi et al. 3 reported a median PFS of 4.9 months in the cisplatin and pemetrexed comparator arm of the large phase III study.
Limitations of the present trial were the reduced patient number in phase II and lack of an arm with checkpoint-inhibitor, and the inclusion of several patients with tumors harboring driver mutations. The last two limitations were caused by the time of study recruitment, when checkpoint-inhibitors were not yet available and tyrosine kinase inhibitors were not fully established as a first-line option. A large phase III trial would be necessary to validate our results. However, due to the currently established new standard of care, every study protocol today would have to include a checkpoint-inhibitor, so that the present trial is unlikely to be repeated in the exact immunotherapy-free schedule as in this manuscript.
In the current COVID-19 era, more frequent visits to hospitals or outpatient practices with d1 and d8 cisplatin schedules may pose an additional challenge to lung cancer patients based on the ongoing corona virus pandemic. This important issue was not present at the time of this clinical trial planning and patient accrual, and it may in fact also become of lesser importance in a hopefully upcoming post-pandemic era in the future.27,28
Our signal-finding trial of split-cisplatin versus standard one day administration of cisplatin and pemetrexed in advanced NsqNSCLC showed comparable ORR rates and OS data but higher compliance to maintenance pemetrexed and resulting longer durations of PFS and TTP in the standard arm. Toxicities between the two schedules differ and the split-dose application schedule is better tolerated in some regards, with a trend toward improved quality of life. Gandhi et al. 3 reported in the Keynote 189 trial a trend to improved OS in the subgroups for cisplatin compared with carboplatin in a combination with pemetrexed and pembrolizumab.
In summary, our findings in this study underline, that split-cisplatin and pemetrexed could be an interesting alternative as partner for checkpoint-inhibitors in an outpatient schedule of the new standard chemoimmunotherapies.
Supplemental Material
sj-pdf-1-tam-10.1177_1758835921996506 – Supplemental material for A randomized, multicenter phase II study comparing efficacy, safety and tolerability of two dosing regimens of cisplatin and pemetrexed in patients with advanced or metastatic non-small-cell lung cancer
Supplemental material, sj-pdf-1-tam-10.1177_1758835921996506 for A randomized, multicenter phase II study comparing efficacy, safety and tolerability of two dosing regimens of cisplatin and pemetrexed in patients with advanced or metastatic non-small-cell lung cancer by Martin Metzenmacher, Hans-Georg Kopp, Frank Griesinger, Niels Reinmuth, Martin Sebastian, Monika Serke, Cornelius Florian Waller, Michael Thomas, Jochen Eggert, Gerald Schmid-Bindert, Mathias Hoiczyk, Daniel Christian Christoph, Martin Kimmich, Burkhard Deuß, Stephanie Seifert, Swantje Held, Martin Schuler, Thomas Herold, Frank Breitenbuecher and Wilfried Ernst Erich Eberhardt in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-pdf-2-tam-10.1177_1758835921996506 – Supplemental material for A randomized, multicenter phase II study comparing efficacy, safety and tolerability of two dosing regimens of cisplatin and pemetrexed in patients with advanced or metastatic non-small-cell lung cancer
Supplemental material, sj-pdf-2-tam-10.1177_1758835921996506 for A randomized, multicenter phase II study comparing efficacy, safety and tolerability of two dosing regimens of cisplatin and pemetrexed in patients with advanced or metastatic non-small-cell lung cancer by Martin Metzenmacher, Hans-Georg Kopp, Frank Griesinger, Niels Reinmuth, Martin Sebastian, Monika Serke, Cornelius Florian Waller, Michael Thomas, Jochen Eggert, Gerald Schmid-Bindert, Mathias Hoiczyk, Daniel Christian Christoph, Martin Kimmich, Burkhard Deuß, Stephanie Seifert, Swantje Held, Martin Schuler, Thomas Herold, Frank Breitenbuecher and Wilfried Ernst Erich Eberhardt in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors thank all the patients and their families for their participation in the study. University Medical Center Essen, West German Cancer Center (WTZ) Department of Medical Oncology served as sponsor of this trial. The “Arbeitsgemeinschaft Internistische Onkologie (AIO)” of the “Deutsche Krebsgesellschaft” accepted the trial as AIO-Trial (AIO-TRK-0212). This study was conducted as a cooperation between the AIO and the German Cancer Consortium (DKTK).
Conflict of interest statement
M.M. reports honoraria for advisory boards from MSD, BMS, Roche, Boehringer Ingelheim, Amgen, Astra Zeneca, Novartis, Pfizer and Takeda
H.G.K. reports no relevant conflicts of interest
F.G. reports research funding to institution from Astra Zeneca, Boehringer Ingelheim, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Siemens, honoraria for educational lectures from Astra Zeneca, Boehringer Ingelheim, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Ariad, Abbvie, Siemens, Tesaro/GSK, Amgen, honoraria for advisory boards from Astra Zeneca, Boehringer Ingelheim, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Ariad, Abbvie, Tesaro/GSK, Siemens, Tesaro, Amgen
N.R. received honoraria for educational lectures and advisory services from Astra Zeneca, Boehringer Ingelheim, BMS, MSD, Pfizer, Roche, and Takeda.
M.S. (Sebastian) reports research funding to institution from Astra Zeneca, honoraria for advisory boards from Astra Zeneca, BMS, MSD/Merck, Roche, Pfizer, Novartis, Boehringer Ingelheim, Takeda, Abbvie, Johnson & Johnson, Amgen, Tesaro, Eli Lilly, honoraria for educational lectures rom Astra Zeneca, BMS, Novartis, Pfizer, Boehringer Ingelheim, Amgen and Eli Lilly.
M.S. (Serke) reports research funding to institution from Astra Zeneca, BMS, Lilly, MSD, Roche, honoraria for educational lectures from Astra Zeneca, Boehringer Ingelheim, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, honoraria for advisory boards from Boehringer Ingelheim, BMS, Lilly, MSD, Pfizer, Roche, Takeda, Abbvie
C.F.W. Honoraria for advisory boards from Astra Zeneca, Boehringer Ingelheim, BMS, Chugai, Pfizer, Roche, Takeda: He received consultancy fees from Mylan; Alvotech; Roche and travel grants from IPSEN, BMS and Lilly.
M.T. reports Honoraria for Scientific Meetings from Astra Zeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Chugai, Lilly, MSD, Novartis Pfizer, Roche, Takeda
Advisory-Board Honoraria from Astra Zeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Lilly, MSD, Novartis, Pfizer, Roche, Takeda
Travelling support from Astra Zeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Chugai, Lilly, MSD, Novartis, Pfizer, Roche, Takeda
Research funding to institution from Astra Zeneca, Bristol-Myers Squibb, Roche, Takeda
J. E. reports no conflict of interest
G. S.-B. reports consultation honoraria from Eli Lilly, Boehringer Ingelheim, Roche, BMS. After completion of study temporarily employed by Eli Lilly from 2015–2017
M. H. Honoraria for advisory boards from Astra Zeneca, Boehringer Ingelheim, BMS, Chugai, Pfizer, Roche and MSD; honoraria for educational lectures from Astra Zeneca, Boehringer Ingelheim and Roche
D.C.C. reports personal fees, non-financial support and other from AstraZeneca, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai, MSD Sharp & Dohme, Novartis, Pfizer, Roche, and Takeda
M.K. reports no conflict of interest
B.D. reports no conflict of interest
S.S. reports no conflict of interest
S.H. reports no conflict of interest
M.S. (Schuler) reports Consultant Honoraria from Astra Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, Novartis, Roche, Takeda.
Honoraria for CME presentations from Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, MSD, Novartis.
Research funding to institution from Astra Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Novartis
T.H. reports no conflict of interest
F.B. reports no conflict of interest
W.E.E. reports research funding to institution from BMS, Astra Zeneca and Eli Lilly, honoraria for advisory boards from Astra Zeneca, BMS, MSD/Merck, Roche, Pfizer, Novartis, Boehringer Ingelheim, Takeda, Abbvie, Bayer, Johnson & Johnson, Amgen, Daichi Sankyo, Eli Lilly, honoraria for educational lectures from Astra Zeneca, BMS, MSD/Merck, Roche, Pfizer, Boehringer Ingelheim, Takeda, Amgen and Eli Lilly.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Elli Lilly Germany [H3E-SB-O069]. Elli Lilly provided financial support for the study to the institution University Hospital Essen. The funding source was not involved in any way in the collection, analysis and interpretation of the data and the submitted manuscript. The University of Duisburg-Essen paid the fee for article processing and open source publication from its own rescources directly to the journal.
Ethics and consent
The study was approved by the ethical institutional review board of the University Hospital Essen, Essen Germany, ID-Number: 12-5106-AF. All patients provided written informed consent before entering any study procedures. The trial was conducted according to Declaration of Helsinki, Good Clinical Practice, and national German and European regulations.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.