
Abstract
Researchers are actively seeking novel targeted therapies for the brain tumour glioblastoma (GBM) as the mean survival is less than 15 months. Here we discuss the proposal that the calcitonin receptor (CT Receptor), expressed in 76–86% of patient biopsies, is expressed by both malignant glioma cells and putative glioma stem cells (GSCs), and therefore represents a potential therapeutic target. Forty-two per cent (42%) of high-grade glioma (HGG; representative of GSCs) cell lines express CT Receptor protein. CT Receptors are widely expressed throughout the life cycle of organisms and in some instances promote apoptosis. Which of the common isoforms of the CT Receptor are predominantly expressed is currently unknown, but a functional response to cell stress of the insert-positive isoform is hypothesised. A model for resistant malignancies is one in which chemotherapy plays a direct role in activating quiescent stem cells for replacement of the tumour tissue hierarchy. The putative role that the CT Receptor plays in maintenance of quiescent cancer stem cells is discussed in view of the activation of the Notch–CT Receptor–collagen V axis in quiescent muscle (satellite) stem cells. The pharmacological CT response profiles of four of the HGG cell lines were reported. Both CT responders and non-responders were sensitive to an immunotoxin based on an anti-CT Receptor antibody. The CALCR mRNA exhibits alternative splicing commonly associated with cancer cells, which could result in the atypical pharmacology exhibited by CT non-responders and an explanation of tumour suppression. Due to the inherent instability of CALCR mRNA, analysis of CT Receptor protein in patient samples will lead to improved data for the expression of CT Receptor in GBM and other cancers, and an understanding of the role and activity of the splice variants. This knowledge will aid the effective targeting of this receptor for treatment of GBM.
Keywords
Introduction
Glioblastoma (multiforme, GBM) is the most common malignant primary brain tumour in adults. Aggressive surgical resection decreases the tumour-cell burden by 99%, leaving about 100 million cells and with cytotoxic adjuvant therapy with an agent such as temozolomide the burden is reduced to about 10 million cells, which would include cancer stem cells. 1 The mean overall survival achieved with wide-local surgical resection, followed by adjuvant therapy of radiation and temozolomide (TMZ) plus maintenance therapy with TMZ, remains only 14.6 months. 2
Glioblastoma (GBM) are highly heterogeneous tumours thought to be derived from oligodendrocyte-type 2-astrocyte (O-2A) progenitors of the astroglial lineage 4 as malignant glioma cells express glial fibrillary acidic protein (GFAP) and respond to similar mitogens and differentiation factors. 5
The tumour architecture (illustrated in Figure 1) includes regions of necrosis and oedema (where the blood–brain barrier is compromised), pro-inflammatory micro-environments with cellular infiltrates and proliferative domains. Vascular-like structures referred to as vascular mimicry have been described. 1 Within these latter hyperplastic structures pericyte precursors1,6 and endothelial cells7–9 share the same genetic modifications as glioma stem-like cells (GSCs), supporting plasticity with regard to differentiation lineages and the existence of dominant GSC clones.3,10–13 It is noteworthy that pericyte precursors are multipotent, 14 are evident in the GBM vasculature in the close vicinity to the zones of proliferation, and a percentage express GFAP as noted in a stroke model. 15 This raises the possibility that re-emerged GBM post-treatment might be derived from the pericyte lineage.

(A) Illustration of the major anatomical features of glioblastomas (GBM); (B) Haematoxylin and eosin stain of a tissue section of GBM with an example of vascular mimicry (central vessel) and oedema; and (C) A serial section showing staining of malignant glioma cells 3 with the anti-human CT Receptor antibody mAb31/01-1H10. Nuclei are stained blue with haematoxylin.
Comprehensive studies have detailed the gene expression profiles of GBM tissue samples10–13 and this is supplemented by an anatomical transcriptional atlas. 16
The current inability to improve or predict patient outcomes based on genetic profiling or histopathological features points to a deficit in our understanding of the driving forces for tumorogenesis of GBM and hence targets for tumour reduction or potential therapy.
GSCs play a role as precursor cells for GBM. 17 They display inherent functional diversity,1,3 convey relative resistance to conventional treatments such as chemo and radiotherapy, 18 and provide invasive potential.19–21 GSCs may also contribute to tumour survival and expansion in a number of hostile (hypoxic, inflammatory) micro-environments. As described for normal stem cell populations, GSCs are likely to be associated with dense vascular beds, 22 which are generally located towards the periphery of GBM, and GSCs are believed to be present within the surrounding neuropil perhaps in a quiescent state. GSCs, also known as brain tumour-propagating cells 19 in contrast to brain tumour-initiating cells,20,23 can be perpetuated and expanded serum free 24 in vitro as high grade glioma (HGG) cell lines.25–27 When formed as xenografts in the brains of immune-compromised mice, HGG cell lines form orthotopic, intracranial (ic) tumours that retain their invasive potential and recapitulate much of the pathology of the original tumour.17,19,20,23,24,27
Some evidence has been presented to support the idea that some GSC populations are normally quiescent (G0 phase of the cell cycle) and re-enter G1 phase for self-renewal, but during quiescence are resistant to radiotherapy and conventional chemotherapy, designed to target proliferating cells. Furthermore, evidence suggests that dying cells targeted by chemotherapy release mitogens that stimulate quiescent stem cells to repopulate the tumour in between cycles of chemotherapy.3,18 Thus, the biology of quiescent stem cells represents further complexity when considering the evaluation of new drugs. GSCs are currently recognised as potential targets for therapy, in which case specific molecular targets on quiescent GSCs should be validated and corresponding therapeutics developed. In support of the idea that quiescent stem cells are responsible for GBM relapse, in childhood lymphoblastic leukaemia, quiescent leukaemic (stem) cells appear to account for relapse-causing minimal residual disease (MRD). 28
In GBM, genetic profiles generated from patient biopsies with considerable cellular hierarchy might not provide enough definition to characterise targets on small subpopulations of quiescent GSCs which have the potential to become dominant clones following expansion after treatment cycles.
In a study that investigated CT Receptor protein expression in a small number of GBM patient biopsies, 86% showed expression that is restricted to glioma cells (example in Figure 2) and smaller cells bearing the GSC associated marker, CD-133. 29 The fact that the surrounding neurophil is negative for CT Receptor 29 in those regions of the brain in which GBM is common suggests CT Receptor is a GBM restricted biomarker. Another report described expression of CALCR mRNA found in 115/152 (76%) of primary tumours 30 as calculated from previous reported data. 12

An example of a Glioblastoma (GBM) tumour stained with an anti-human CT Receptor monoclonal antibody mAb30/07-9B4. Malignant glioma cells are stained brown and nuclei are stained blue with haematoxylin. 29
The widespread expression of the calcitonin receptor (CT Receptor)
CT Receptor isoforms are expressed in a wide range of tissues throughout the life cycle of mammals, under conditions such as cell stress, inflammation and wound healing, and in a range of diseases (Table 1). 31 In spite of this wide expression, it is not normally expressed in the cerebrum/cortex where GBMs typically arise but is restricted to specific neuronal networks in the limbic system, and in the mid and hindbrain.
Updated and abbreviated list of tissues that express CTR from several mammalian species. A more comprehensive list with references is listed in Wookey et al. 31
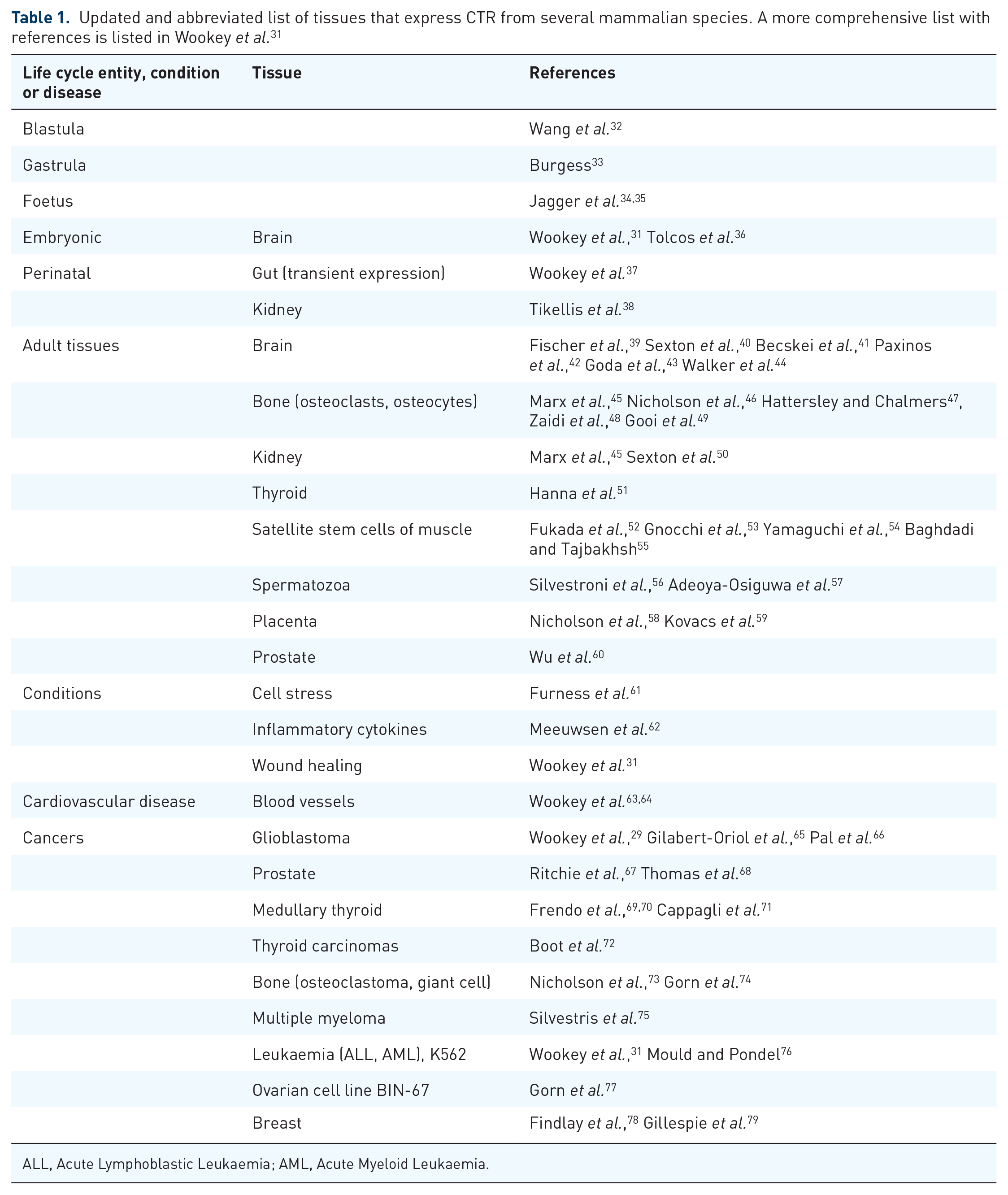
ALL, Acute Lymphoblastic Leukaemia; AML, Acute Myeloid Leukaemia.
There are two common isoforms, CT Receptora, insert-negative and CT Receptorb insert-positive. 80 The insert positive form was originally isolated from a breast cancer cell line and has an additional 16-amino acid sequence within the first intracellular loop. The insert negative form has the more extensively characterised pharmacology and appears to be the relevant isoform for the well-defined physiology of calcitonin signaling and calcium metabolism.
Data from transfected COS (monkey kidney) cell lines that express either of the two human isoforms show CT Receptora is located predominantly in the plasma membrane (Figure 3) whereas CT Receptorb is largely intracellular, located in the perinuclear domain presumably in small membranous elements and has a lower molecular weight suggesting an unglycosylated form that has not been processed for normal cell-surface expression. 81 In transfected cell lines flow cytometry experiments confirm that lesser amounts of CT Receptorb are found on the plasma membrane, but the relative distribution is cell line dependent. 82

COS-7 stable transfectants expressing CT Receptora and CT Receptorb 81 stained with primary anti-human CT Receptor antibody mAb31/01-1H10, secondary goat anti-mouse IgG2a:AF568 and imaged using confocal microscopy (Zeiss LSM 800). Nuclei are stained blue with DAPI (4’,6 diamidino-2-phenylindole).
The CT Receptorb isoform is expressed more widely than previously thought across the mammalian order. In the majority of mammalian species examined so far (exception Muroidae), the position of insertion is identical at the beginning of the second transmembrane span; however, the insertion varies in amino acid sequence and length (16–18), and would be predicted to interfere with the interaction of the receptor with its primary transducers. The recent description of the widespread expression of insert-positive isoforms throughout the mammalian species does, however, suggest a specific function of CT Receptorb in cell physiology. 81
There are no conclusive data yet that demonstrates in which normal and diseased tissues CT Receptorb is more highly expressed and what its function might be. However, there is some evidence that this isoform is more highly expressed in ovary and placenta 80 and CT Receptorb mRNA predominates in a group of samples of mononuclear blood cells. 83 The recent validation of a mouse monoclonal anti-human CT Receptorb antibody 81 will aid in the resolution of questions about expression in normal tissues, during inflammation and diseases. Unpublished data from our group on the expression of CT Receptorb upregulated in cell stress (see also Adeoya-Osiguwa and Fraser) 57 are consistent with a cytosolic function.
Pharmacology of CT Receptora and CT Receptorb
The pharmacology of these isoforms is quite different in terms of the second messenger outputs which, for CT Receptora, includes adenylyl cyclase activation, phosphorylation of ERK1/2 and p38 MAP kinase, as well as mobilisation of intracellular calcium (summarised in Figure 4 below). This might be expected given the location of the insert in relation to the binding of CT Receptor to its primary transducer. 84 In heterologous systems (COS-7 and HEK 293), second messenger coupling of CT Receptorb is substantially reduced: the potency for adenylate cyclase activation is more than 100-fold weaker, ligand stimulated phosphorylation of ERK1/2 is reduced in its maximum with some reduction in potency and CT Receptor-dependent mobilisation of intracellular calcium is undetectable. 82 Such outputs are also cell type dependent82,85 and mediated by different G protein complexes. However, it was noted that stimulation with salmon CT of HEK-293 transfectants (both CT Receptor isoforms compared to vector control) resulted in acidification of the media 86 suggesting possible metabolic outputs or involvement in efflux mechanisms from acidified intracellular compartments.

Typical signaling pathways from CT Receptora and CT Receptorb.
The CT Receptor ligand promoted conformation of Receptor bound G protein complexes influences their residency on the plasma membrane and contributes to ligand-mediated biased agonism of CT Receptora. 87 However, little is known about these mechanisms in the context of insert-positive CT Receptorb and how the peptide insert might perturb residency, and thus coupling of G protein complexes.
CT Receptor protein was detected in the 5/12 (42%) of HGG cell lines (isolated from human biopsies) tested, 27 including JK2 (mesenchymal), PB1 (proneural/classic), SB2b (mesenchymal/classic) and WK1 (classic), using immunoblotting with the monoclonal antibody mAb31/01-1H10 30 which binds an intracellular epitope. In contrast to the major, broad CT Receptor band of 70–80 kD from COS-7/CT Receptora transfectants, 81 the upper band is tight, with an apparent molecular weight (MW) of approximately 57 kD, consistent with an unglycosylated form. Unglycosylated CT Receptor is largely confined to the cytosolic domain and is characteristic of the CT Receptorb isoform 81 (Figure 3 above). However, as discussed above a small proportion of CT Receptorb is located on the cell surface, which is cell line dependent, 82 and in the case of HGG cell lines JK2, SB2b and WK1, CT Receptor is found in the membrane fraction determined from immunoblot. 30 In PB1 there is much less CT Receptor protein found in the membrane fraction. Rapid turnover of unglycosylated human CT Receptora protein located in intracellular compartments has been described. 88 Further studies including nanopore sequencing to identify splicing events will be important to improve our understanding of the biology and functional consequences of these observations.
The pharmacology of CT Receptor was studied in these HGG lines 30 and only SB2b had functional CT Receptor as determined by responses to classic ligands which stimulated adenylyl cyclase, calcium mobilisation and ERK1/2 phosphorylation. Pharmacological studies are discussed in more detail below.
Hypothesis on the mechanism of the CT Receptorb isoform
This hypothesis is built on several separate ideas. Firstly, intracellular CT Receptors are folded in the membrane of intracellular structures attached to the cytoskeleton. 89 Secondly, unglycosylated CT Receptorb is confined to the cytosol (Figure 3 above) and is concentrated in a structure thought to be the microtubule organising centre (MTOC). In experiments similar to those described by Safaei et al. 90 we found cytotoxin concentrated in the exosomes harvested from MG63 cells treated with staurosporine and these exosomes were also positive for CT Receptor (unpublished results). As these exosomes originate from lysosomes (pH 3–4) their release is likely to contribute to acidification of the media as described previously 86 for CT Receptorb.
The upregulation of CALCR mRNA in response to cytokines TNFα and IL1β by primary cultures of human astrocytes has been described, 62 demonstrating a response to cell stress. 61 Furthermore, in U87 MG cells treated with staurosporine, nanopore sequencing of long cDNA products from CT Receptor mRNA has established alternative splicing events that include exon 10 such that the CT Receptorb isoform (insert-positive) is significantly upregulated, 81 although total CALCR mRNA remains unchanged.
Taken together there is evidence for the upregulation of CT Receptorb in the cytosol of stressed cells and the production of CT Receptor-positive exosomes laden with cytotoxins, which as hypothesised, amounts to new important mechanism together with a number of other cellular stress responses.
In the context of the expression of CT Receptor by malignant glioma cells and GSCs, targeting CT Receptor might provide an opportunity to overcome resistance to chemotherapeutics and treat the pool of GSCs thought to be responsible for relapse.
Calcitonin/CT Receptor, cell survival/apoptosis and the cell cycle
There are several reports that describe data showing CT or CT Receptor promote proliferation/survival or apoptosis resulting in decreased survival in model cancer cell lines. The effect of CT Receptor expression and calcitonin-dependent CT Receptor activation differs according to the cell line under investigation.
CT stimulates proliferation early in treatment protocol of T47D cells (derived from human breast cancer) and then later inhibits proliferation in the log phase. 91 In serum deprived LLC-PK cells, derived from porcine kidney, CT reduced cell survival 92 perhaps by induction of apoptosis.
In a study of serum-starved transfected HEK-293 cells induced to express either human hCT Receptora compared to hCT Receptorb or vector alone, treatment of these lines with salmon CT resulted in decreased proliferation, in the accumulation of cells in G2 phase of the cell cycle in the hCT Receptora transfectant but not the hCT Receptorb transfectant. 86 In the hCT Receptora transfectant, ERK1/2 activation mediates modulation of the cell cycle via p21Cip1. However, G2-arrest was unexpected as p21Cip has been shown to inhibit CDK2 rather than CDK1 (cdc 2) 93 and results in a cycle block at G1.94,95 This anomaly has never been resolved as far as we are aware and raises questions about the validity of CT Receptor transfectants to probe the real biological functions of CT Receptor.
In a subsequent report CT induced apoptosis in an hCT Receptora transfectant under conditions of low serum which were not observed with replete serum. 96 While one explanation might be a factor present in serum that overcomes the CT effect, another possibility is metabolic reprogramming driven by starvation and cell stress associated with autophagy which is independent of caspase 3. 96 In this regard, in a p53–/– mouse model with thymic lymphoma, CT Receptor is essential for the transmission of the effects of amylin for metabolic reprogramming, induction of apoptosis and tumour regression. 97
SiRNA mediated knockdown of CT Receptor in TT2609-C02 cells derived from follicular thyroid carcinoma, resulted in G1 arrest, a decrease in proliferation and an increase in caspase 3 activity. 72 The authors conclude that CT Receptor is one of several genes important for survival of thyroid carcinomas and therefore can be classified as a putative oncogene.
In human prostate cancer cell lines and mouse models, CT and CT Receptor activate survival of cells following cytotoxic insult 68 and enhanced tumour growth 98 consistent with the induction of apoptosis following knockdown of CT Receptor. 99
Consistent with these results are further reports that CT prevents apoptosis in osteocytes and osteoblasts 100 and promotes survival of osteoclasts. 101
Overall in the context of different cell lines maintained in normal serum (unstarved), CT/CT Receptor promotes cell survival and proliferation, while promotion of apoptosis is associated with P53 status, metabolic reprogramming and nutrient deprivation.
Stem cell quiescence and a role for a surrogate ligand of CT Receptor
In skeletal muscle stem (satellite) cells maintenance of quiescence is dependent on activation of CT Receptor 54 and it is now proposed that an active cell autonomous Notch–Collagen V (COL V)–CT Receptor axis maintains the quiescent muscle stem cells in their niche. 55 Notch also contributes to the stem-like character of glioma cells. 102
The mechanism of how COL V acts as a surrogate ligand of CT Receptor is yet to be described. We have modelled [unpublished] the carboxy cleaved peptide of collagen V and found similarities when arranged as a 310 helix with salmon CT. In this case the carboxyl tail of nascent COL V might act as a surrogate ligand for CT Receptor after it is cleaved. However, the researchers 55 used a commercial collagen V isolated from human placenta and it is unclear whether this is pure mature collagen V or contains pre-pro-collagen V and processed peptides. 55 If this work can be corroborated this discovery will have a profound effect on our understanding of CT Receptor as expressed in the context of many tissues. As presented in Table 1, there is a wide expression of CT Receptor throughout the life cycle in many tissues, sometimes expression is transient and sometimes persistent, and expression is found in cells that play a prominent role in a range of diseases.
For instance, this axis might well be a driving factor in atherosclerosis in which we have identified expression of CT Receptor63,64,103,104 and expression of collagen V is also known.103,104
Formation of granulation tissue is a histological hallmark of wound healing following tissue injury. 105 COLV immunostaining predominates in the blood vessel walls of the granulation tissue and COLV transcript is expressed in the fibroblast-like cells of the granulation tissue.105,106 Similar cells and blood vessels express CT Receptor in a mouse model of wound healing. 31
Genotyped profiles of GBM
Extensive genetic profiles of GBM patient biopsies have been published,10–12 including regions of chromosomal aberrations both broad and focal 10 together with the somatic genomic landscape. 12
Gliomas with broad amplification of chromosome 7 have properties different from those with overlapping focal EGFR amplification (chr7p11) and the broad events around chr7q32 (CALCR gene location chr 7q21.3) which appear to act in part through effects on MET (CXCR4) and its ligand HGF and correlate with MET dependence in vitro. 10 It should be noted that CALCR maps close to MET.
Alternative splicing has also been recognised as a driving force of tumorogenesis. 107 Aberrant profiles observed in tumours mostly reflect the selection of endogenous alternative splicing variants with different functional properties that allow the malignant progression of initiated tumour cells. Selected functions relate, for example, to sustained proliferation, evasion of apoptosis, metabolic adaptation, or angiogenesis. 108 Results from our group investigating the U87 MG cell line have demonstrated increased frequency of exon 10 (insert-positive sequence) splicing-in when these cells are stressed with cytotoxin treatment. 81 The possibility of other splicing events that might result in receptor inactivation are currently under investigation.
In a study on oncocytic thyroid carcinomas 72 hemi-methylation in the region of the 5’UTR of CALCR was characterised, upregulation of CALCR mRNA was found in the majority of samples and in the cell line TT2609-C02 reduction in the expression of CTR leads to a pause in the cell cycle, upregulation of caspase 3 mRNA and enhanced cell death. Consistently, there was no loss of heterozygosity of chromosome 7 which was interpreted as a role for maternal and paternal genes imprinted on this chromosome including CALCR. These data support a role for CT Receptor as an oncogene.
Regulation of CALCR transcription and stability of mRNA
The structure of human CALCR gene has been described in BIN-67 cells (human ovarian carcinoma) cell line, 77 human osteoclasts and MCF-7 cells (human breast adenocarcinoma). 109 Promoters P1 and P2 were demonstrated in transfected T47D cells (human breast cancer)110,111 and a further promoter (POc) is specifically active in human osteoclasts. 112 This paper also described the tissue-specific splicing of the CALCR 5’UTR and the transcripts that are generated. Analysis of the 5’UTR of human CALCR reveals multiple Sp1 binding sites 113 and CpG islands. The regulation by Sp1 is of particular interest because many genes upregulated during stress are also regulated by this transcription factor which has been proposed to drive the adaptive response of cancer cells to hypoxia. 114
The structure of the murine CALCR gene from brain has been determined 115 and later CALCR mRNA shown to be transcribed from three promoters (P1, P2 and P3) of which P3 is osteoclast specific and the promoters become functional through alternative splicing of exons in the 5’UTR. 116 A comparison of the exon structures in human and mouse CALCR has been published. 112 The structure of porcine CALCR gene has been reported from LLC-PK1 cells. 117
In the 3’UTR of human CALCR mRNA there are eight AUUUA sequences and five in porcine CALCR. 118 These sequences have been demonstrated to increase the instability of transcripts.119,120 CALCR mRNA levels are generally low in tissues and HGG cell lines 30 perhaps reflecting the inherent instability of this mRNA due to multiple (AUUUA) sequences. This instability could explain the low representation of CALCR mRNA in some genetic studies of GBM patients. 11
Targeting inactivated CT Receptor in GBM
The pharmacology of responses to CT ligands was reported in these four HGG cell lines. 30 Only SB2b responded to CT-like ligands (human CT, salmon CT, rat amylin) indicating that CT Receptor is non-functional in the cell lines JK2, PB1 and WK1. It is likely that this inactivation results from alternative splicing common in cancers including the possibility that expression of CT Receptorb predominates. A conclusion drawn from this study is that pharmacological intervention of CT Receptor is unlikely to provide an avenue for treatment of GBM.
Our group 65 has published a study with HGG cell lines which characterises the potency of an anti-hCT Receptor antibody conjugated to cytotoxins monomethyl-aurostatin E (MMAE) or the plant toxins diathin-30 121 and gelonin (ribosome-inactivating proteins). The anti-hCT Receptor antibody deployed in the ADC or immunotoxin binds an extracellular epitope and is internalised. The potency of the immunotoxin (EC50 10–20 pM) is greatly increased with saponins (triterpene glycoside SO1861) which enhance the release of the toxin from the acidic lysosomes. 65
Three of the HGG cell lines (JK2, SB2b, WK1, all classic/mesenchymal) 27 were equally sensitive to the immunotoxin and expressed high levels of the CT Receptor protein in the membrane fraction as determined by immunoblot 30 using an anti-CT Receptor antibody that recognises an intracellular epitope. Although all four HGG cell lines displayed similar levels of CT Receptor using whole cell lysates, 65 one HGG cell line PB1 (proneural) expresses low levels of CT Receptor in the membrane fraction 30 and is relatively resistant to the immunotoxin. 65
These data suggest that a potential treatment of GBM stem-like cells is possible using immunotoxin directed against CT Receptor regardless of the pharmacological status of the receptor.
The proposed mechanism 65 of this therapy (refer to Figure 5) involves binding to the extracellular domain of CT Receptor on the plasma membrane, uptake of the immunotoxin via the endosomes into the lysosomes (with low pH) and cleavage of the immunotoxin (either protease or pH mediated depending on the linker). The escape of the toxin from the lysosomal compartment 65 is enhanced by the triterpene glycoside SO1861. 122 The toxin targets the ribosomes. 121
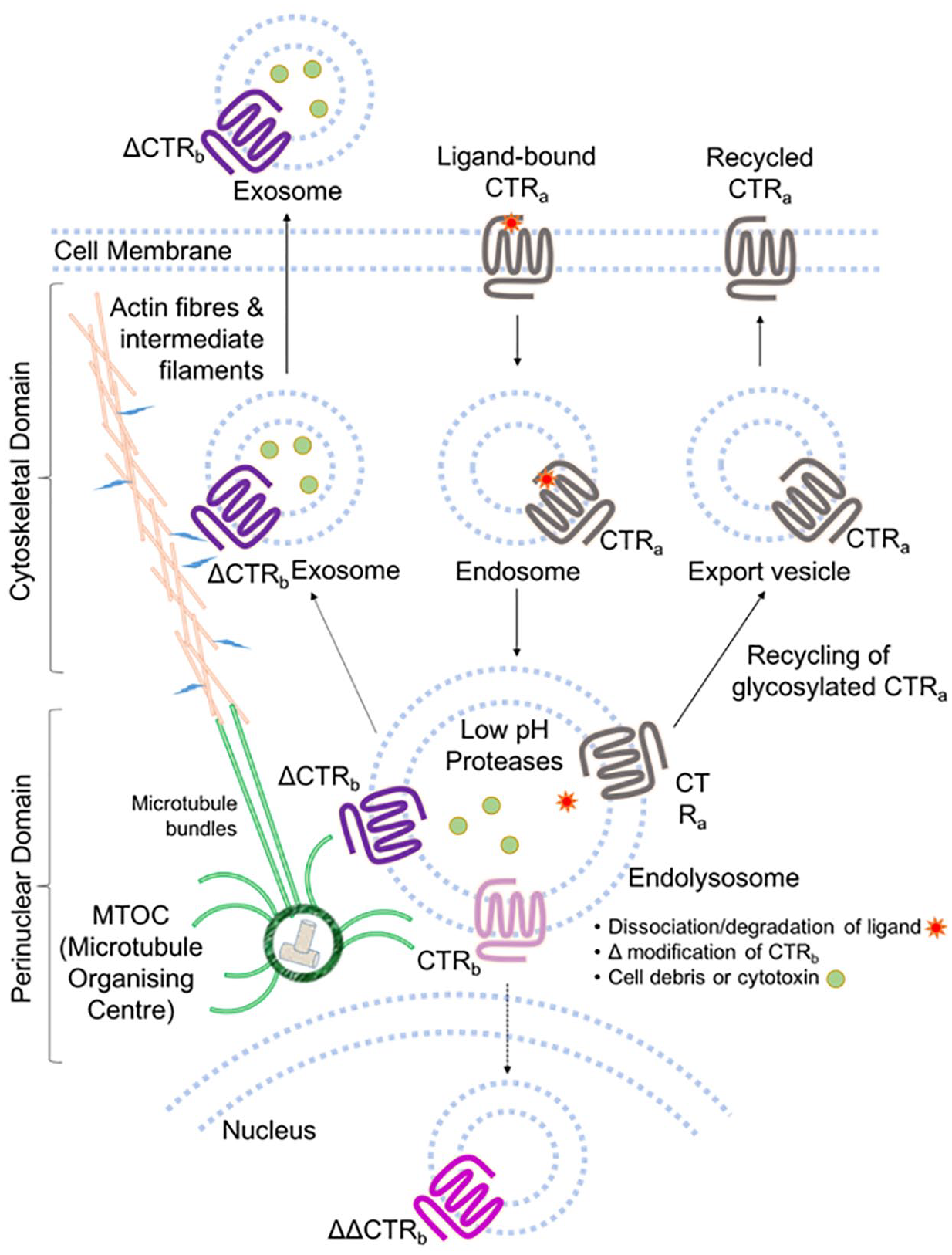
Proposed mechanism of CT Receptorb in the excretion of cell debris and cytotoxins via exosomes.
Whether the HGG cell lines express CT Receptora, CT Receptorb or an alternatively spliced isoform is yet to be established. From our current analysis elevated CT Receptor expression in the membrane fraction is necessary for sensitivity to the immunotoxin as PB1 is deficient in this respect and is resistant to the immunotoxin.
Animal studies are planned to test the immunotoxins in vivo and further refinements of the prototype immunotoxin, to improve tumour penetration, are also in progress.
Evidence for CTR as a tumour suppressor and/or an oncogene
Tumour suppressor genes regulate a diverse range of cellular activities including cell cycle checkpoint responses and mitogenic signaling. 123 Classic tumour suppressors have three principal characteristics, firstly they are recessive and undergo biallelic inactivation in tumours, secondly inheritance of a mutant allele potentiates tumour initiation, and thirdly, the same gene is frequently inactivated in sporadic tumours. 123
CT Receptor has been shown to influence the cell cycle with induction of quiescence (G0) for satellite stem cells and checkpoint G1 for cell lines as discussed above. Two reports have identified inactivated CT Receptor expressed in GBM.30,66 As CT Receptor is upregulated with cell stress 61 and ligand activation results in apoptosis (discussed above) then inactivation of CT Receptor by mutation or alternative splicing (U87 MG, discussed above) could result in survival of tumour cells. It should be noted at this point that U87 MG might not have originated from GBM. 124 CT Receptor protein is expressed by HGG cell lines in which CT Receptor activation is either non-canonical or inhibited by mutation or deletion, or the CT Receptorb isoform is preferentially expressed.
Pal et al. 66 describe the downregulated levels of CALCR transcripts in GBMs from data sets TCGA, GSE7696 and the Indian cohorts. However, as described above, low levels might be expected to result as the transcripts include eight repeat AUUUA sequences in the 3’UTR that are responsible for instability. The steady state levels are not an indication of the rates of transcription. They also found altered activity of mutated CT Receptor derived from a small cohort 66 of GBM patients and drew conclusions about survival compared to normal CT Receptor. While much of the data are not conclusive, the idea that CT Receptor might act as a tumour suppressor and/or oncogene warrants further exploration.
Evidence for a role as an oncogene includes knockdown of CT Receptor in tumour cell lines as outlined above, which results in apoptosis and suggests, in the case of mutant CT Receptor, a further function of ligand-insensitive CT Receptor for tumour survival as proposed in Figure 5. It remains to be demonstrated that knockdown of inactivated (mutant) CT Receptor leads to cell death in the case of GBM.
Conclusion
CT Receptor is a G protein-coupled receptor (GPCR) that is highly expressed in patient biopsies between 76% (CALCR mRNA) and 86% (CT Receptor protein). Furthermore, it is expressed by malignant glioma cells, in cells that express markers of brain tumour initiating cells and 42% of HGG cell lines that represent tumour stem cells.
Quiescent cancer stem cells represent minimal resistant disease and enter the cell cycle in response to treatment to re-establish tumour malignancy. We postulate that the maintenance of quiescence results from activation of the Notch–COL V-CT Receptor axis as has been proposed for skeletal muscle satellite (stem) cells.
There are likely to be several potential mechanisms for the upregulation of CT Receptor in GBM. Firstly, the region around chromosome 7q21.3 (CALCR gene) is frequently amplified. Secondly, the transcription factor Sp1 regulates genes important for stress responses in many tissues including GBM and Sp1 has been shown to stimulate CALCR mRNA transcription in cell lines.
The potential role of the CT Receptorb isoform is discussed in a mechanism that provides resistance to cytotoxins and drug resistance in glioblastoma and possibly other cancers.
In HGG cell lines CT Receptor is frequently pharmacologically inactive although the protein is detected by immunoblotting. Inactivation might result either by inactivating mutations/deletions/insertions or alternative splicing resulting in inactivation or a switch to the CT Receptorb isoform. The inactivation of CT Receptor would be consistent with its role as a tumour suppressor. Furthermore, knockdown of CT Receptor promotes apoptosis consistent with a role as an oncogene.
The antibody (mAb2C4) that binds the extracellular epitope of human CT Receptor has been developed as an immunotoxin to study its potency with HGG cell lines. The mAb2C4:dianthin immunotoxin has an effective EC50 of 10–20pM as compared to an equivalent ADC (mAb2C4:MMAE) which is 250 times less potent.
Targeting CT Receptor expressed by quiescent cancer stem cells with the immunotoxin is expected to provide an additional weapon, in combination with traditional therapy that targets dividing malignant glioma cells, for the treatment of glioblastoma. This novel therapy aims to eradicate minimal residual disease associated with this cancer.
Footnotes
Acknowledgements
The authors would like to thank Apop Biosciences Pty Ltd for access to the mAb2C4 antibody (CalRexin).
Conflict of interest statement
PJW and DLH own shares in Apop Biosciences (formerly Apop Imaging) which owns intellectual property around the anti-CTR antibodies mAb2C4 and mAb9B4. The other authors declare no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: SGBF is an ARC Future Fellow (FT180100543).