Introduction
Worldwide, tumours are currently one of the main causes of death with approximately 9.6 million deaths a year and an incidence of approximately 18.1 million new cases in 2018.
1
Consequently, the prevention, diagnosis and treatment of cancer represent, more than ever before, goals towards which all international administrations are moving; however, the results are not always tangible and comforting. One of the causes of these total or partial failures has been identified as the absence of adequate nationwide awareness programmes regarding the lifestyles most likely to reduce the incidence of cancer, as well as a shortage of funding for research and for the distribution of the most innovative medicinal products to the whole population. On the other hand, cancer is a huge business opportunity for pharmaceutical companies around the world, even more now than in the past. A review by Hong and colleagues
2
shows that in the United States between 2011 and 2016, spending on cancer drugs grew by over 50% (from US$26.8 to 42.1 billion). Although the introduction of immune checkpoint inhibitors has dramatically changed the treatment of a number of cancer types, they bring a host of new adverse effects to be managed and a further exponential increase in both direct and indirect costs. Researchers are exploring new strategies making it possible to deal in the years to come with what can be defined as an authentic politico-socio-economic emergency that is now well identified, also regarding the possibly over simplistic terminology such as the ‘financial toxicity’ of cancer treatments. The price, not merely in economic terms, that is likely to have to be paid is dramatic meaning that much of the world population, even in higher income countries, could be excluded from access to the most novel cancer treatments. One well consolidated, albeit far from decisive, pathway undertaken in an attempt to reduce costs is the authorization of biosimilars and generic medicines. Another option to be given careful attention is that of reconsidering (at equal efficacy) the authorized doses of certain medicinal products, especially those with higher costs, particularly when used on a large scale such as those indicated for the treatment of the most frequently terminal cancers.
A few examples of more or less recent oncological agents that have undergone changes to their authorized regimens or that, in clinical practice, are almost always administered using posologies that differ from those originally authorized are reported in the following.
Abiraterone Acetate (AA)
administered at a once-daily dose of 1000 mg on an empty stomach is the current standard of care for the treatment of hormone sensitive metastatic prostate cancer.3–6 An interesting article published recently in the Journal of Clinical Oncology based on the hypothesis that administering a dose of AA equal to one-quarter (250 mg) of the standard dose with a light, low fat breakfast is equally efficacious, showed its noninferiority in terms of both prostate-specific antigen response and time to progression compared with the authorized dose. The incidence of side effects was also similar.
7
This finding could pave the way for the exploration of other alternative regimens in which AA is administered with food, such as a dose of 500 mg every other day, or even once every 4 days. It would appear unnecessary to stress the significant pharmacoeconomic implications that could be derived from this result.
Niraparib (N)
is a poly (adenosine diphosphate-ribose) polymerase inhibitor approved in the USA and Europe for maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum based chemotherapy. In the pivotal study, N was administered at a once-daily dose of 300 mg.
8
However, approximately 70% of patients had to reduce this dose due to adverse events and about 15% had to discontinue treatment, mainly for grade >3 thrombocytopenia. The retrospective study conducted by Berek and colleagues showed that, after adjustments to the dose of N, 200 mg is the dose most often used. Their analysis concluded that in patients weighing <77 kg (presumably the majority) or with platelet values <150,000 mm3 at baseline, N administered at a dose of 200 mg is able to achieve the same results, but with a far more acceptable toxicity profile.
9
Lapatinib (L)
is another example of how food is able to significantly change the bioavailability of a medicinal product. L was approved by the Food and Drug Administration (FDA) in 2007 in combination with capecitabine for advanced HER2-positive breast cancer in progression after previous treatments including anthracyclines, taxanes and trastuzumab. The regimen envisaged administering L at a flat dose of 1250 mg a day an hour before or an hour after breakfast, continuously.
10
However, when L is administered with food, the (geometric) mean increase for the area under the concentration–time curve was 167% for low fat meals and 325% for high fat meals. These results are not surprising, given that food often increases a drug’s bioavailability, thereby making it possible to administer a drug at lower doses.
11
Authors have gone so far as to make the provocative suggestion that L could be administered with food at 1/5 of the standard dose (250 mg instead of 1250 mg) and could be taken with a glass of grapefruit juice.
12
Regorafenib
is currently indicated for the treatment of metastatic colorectal cancer after at least two lines of therapy,
13
for gastrointestinal stromal tumours (GIST) following progression on treatment with imatinib and sunitinib, and for hepatocellular cancer following progression on treatment with sorafenib, at a dose of 160 mg a day for 3 weeks, followed by a 1-week break in 28-day cycles.
14
Gastrointestinal (GI), skin and hepatic toxicity often make it necessary to reduce the dose or even discontinue the treatment in heavily pretreated patients. The reDOS trial showed that a dose escalation of 40 mg a week starting from a dose of 80 mg makes it possible to limit toxicity, whilst maintaining efficacy, with an improvement in overall survival in the investigational arm.
15
Ceritinib (C)
750 mg fasted is approved for treatment of patients with ALK receptor tyrosine kinase gene (ALK)-rearranged (ALK-positive) non-small cell lung cancer (NSCLC) previously treated with crizotinib.
16
In an attempt to reduce the entity of gastrointestinal toxicity whilst maintaining the same pharmacokinetic and efficacy profile, part one of the ASCEND-8 study determined whether administering C 450 mg or 600 mg with a low fat meal can enhance GI tolerability versus 750 mg fasted in patients with ALK-positive NSCLC while maintaining similar exposure.
17
This study demonstrated that C 450 mg administered with food has a similar exposure to the medicinal product and a GI toxicity profile that is considerably more favourable than C 750 mg in fasted patients with ALK-positive NSCLC. On the other hand, with the 600 mg dose, again administered with food, the steady state pharmacokinetics showed 25% higher levels, and it would therefore not appear to be suited to the aim of limiting toxicity.
Sunitinib
is currently indicated for GISTs that are refractory to imatinib.
18
and metastatic renal cell carcinoma (MRCC) with a recommended starting dose of 50 mg to be administered orally once daily, for four consecutive weeks, followed by a 2-week break (4/2 regimen) in cycles with an overall duration of 6 weeks.
19
It is also indicated for the treatment of pancreatic neuroendocrine tumours; however, in this case, the recommended dose is a once-daily oral administration of 37.5 mg, without break periods.
20
However, in GISTs and MRCC, following the considerable toxicities reported that usually force approximately 50% of patients to reduce the starting dose, in recent years, clinicians have increasingly opted for an alternative regimen at the same daily dose, but administered for 2 weeks followed by a week’s break (2/1 regimen). In the study by Bracarda and colleagues in MRCC, the shift due to toxicity from the 4/2 regimen to the 2/1 regimen led to a reduction in >grade 3 toxicity from 45.7% to 8.2%. More specifically, fatigue, hypertension, hand-foot syndrome and thrombocytopenia were less frequent.
21
However, recently a panel of experts ruled that in MRCC, due to the methodological limits of the studies considered and, therefore, in the absence of certain efficacy data for the 2/1 regimen, despite the better tolerability observed it is still preferable to start therapy with the standard 4/2 regimen.
22
Vinorelbine (V)
is administered in the treatment of advanced breast cancer after failure of standard therapy, as a single agent or in combination, and as a first-line treatment for advanced NSCLC, as a single agent or in combination. When used as both a single agent and in combination, V is currently usually administered at a dose of 25–30 mg/m2 on days 1 and 8 of 21-day cycles; however, in the original authorized regimen, which dates back some 25 years, this agent was administered at the once-weekly dose of 30 mg/m2 continuously until disease progression or unacceptable toxicity.23,24
Capecitabine
is currently indicated as adjuvant therapy for stage III bowel cancer, for the treatment of metastatic colorectal cancer, as a first-line therapy for advanced gastric cancer in combination with a regimen containing platinum, in combination with docetaxel for the treatment of patients with locally advanced or metastatic breast cancer following the failure of a chemotherapy regimen containing an anthracycline, as monotherapy for the treatment of patients with locally advanced or metastatic breast cancer following the failure of a chemotherapy regimen containing taxanes and an anthracycline, or for which further anthracycline therapy is not indicated. When used as a single agent, the recommended starting dose is 2500 mg/m2/day in two oral administrations on a full stomach (breakfast and dinner) for 14 days followed by a 1-week break, in 21-day cycles. In combination therapy, the recommended daily dose is 1600–2000 mg/m2/day, or 1250 mg/m2/day when administered continuously, as in the case of concomitant radiotherapy or metronomic chemotherapy. Two doses of capecitabine are currently available on the market: the 150 mg tablets (very rarely used) and the 500 mg tablets, which are practically the only kind used in clinical practice. This now consolidated habit by clinicians of using the 500 mg formulation alone, has led to approximations (often rounding down) of the overall amount of capecitabine that should be administered.25,26
Bevacizumab
is an antivascular endothelial growth factor monoclonal antibody indicated in combination with a number of chemotherapy agents for a broad spectrum of advanced tumours (ovarian, breast, lung, cervical, renal and colorectal cancers). For example, in the treatment of colorectal cancer, in which this medicinal product is extensively used, the recommended dose of bevacizumab, is 5 mg/kg or 10 mg/kg once every other week, or 7.5 mg/kg or 15 mg/kg of body weight once every 3 weeks, depending on the regimen used.27–30 The doses most frequently used in clinical practice are 5 mg/kg every other week and 7.5 mg/kg every 3 weeks.
Cabazitaxel (Cab)
in combination with prednisone or prednisolone is indicated for the treatment of patients with castration-resistant metastatic prostate cancer who have previously been treated with a regimen containing docetaxel. The recommended dose is 25 mg/m2 administered every 3 weeks.
31
However, one recently published phase III study showed the noninferiority of Cab at a 20% lower dose with an approximately 15% lower incidence of serious adverse events (primarily medullary toxicity, febrile neutropenia and diarrhoea).
32
Docetaxel (D)
is approved as a single agent (100 mg/m2 q21),33,34 or in combination in the adjuvant treatment of breast cancer,
35
in advanced breast cancer36–38 and as a second-line therapy for advanced NSCLC (75 mg/m2 q21)39,40 in combination with prednisone. It is also approved in the treatment of patients with metastatic prostate cancer (75 mg/m2 q21)
41
in combination with cisplatin and 5-fluorouracil for the treatment of patients with metastatic cancer of the stomach and gastro–oesophageal junction who have not received prior treatment for the metastatic disease
42
and, finally, again in combination with cisplatin and 5-fluorouracil, for the induction treatment of patients with locally advanced squamous cell carcinoma of the head and neck.43–45 Nevertheless, in clinical practice, in order to minimize toxicity, docetaxel is now also often administered at a dose of 60 mg/m2 q21,
46
or with the weekly regimen of 30–40 mg/m2/w for six consecutive weeks every 8 weeks47,48 or even with a biweekly regimen of 50 mg/m2.
49
Liposomal doxorubicin
is approved for the treatment of ovarian cancer, breast cancer, multiple myeloma, and Kaposi sarcoma.50–52 In monochemotherapy it is still authorized at a dose of 50 mg/m2 every 28 days; however, in clinical practice the dose that is usually administered is 20% lower (40 mg/m2), with a significant reduction in the incidence and severity of palmar-plantar erythrodysesthesia and mucosites.
53
Nivolumab
, an anti-PD-1 monoclonal antibody, represents the most recent example of how an authorized dose (3 mg/kg every other week) can be altered without being based on a solid scientific rationale, but for primarily financial reasons. Nivolumab is indicated for the treatment of a number of solid tumours such as melanoma, renal cell carcinoma, NSCLC, urothelial cancer, squamous cell carcinoma of the head and neck, hepatocellular carcinoma, colorectal carcinoma and in haematological settings in Hodgkin’s lymphoma.54–62 The strange thing is that since its marketing authorization, there has been a switch from using a dose of 3 mg/kg to a flat dose every 2 or 4 weeks (Table 1) thanks to debatable studies able to demonstrate an equivalence of efficacy.63,64 A recent editorial by Ratain and Goldstein
65
states that there is significant pharmacokinetic and pharmacodynamic evidence supporting the theory that nivolumab can work just as well, and probably with a better immune-correlated toxicity profile, even at considerably lower doses. In a phase I study in 2012, nivolumab at 0.1 mg /kg (about 3% of 3 mg/kg) every other week showed activity and the ability to saturate receptors.66,67 This means that there is most likely no dose–response correlation and that nivolumab could probably be administered at doses considerably lower than those currently used without prejudicing the results.
H&N, Head & Neck; HD, Hodgkin Disease; NSCLC, nonsmall cell lung cancer; RCC, renal cell carcinoma; SCC, Squamous Cell Carcinoma.
The same thing would appear to apply for pembrolizumab, another anti-PD-1 monoclonal antibody.
68
For example, pembrolizumab monotherapy is currently administered at a flat dose of 200 mg every 3 weeks as first-line therapy for advanced NSCLC, in classic Hodgkin’s lymphoma and in urothelial cancer69–71 and at 2 mg/kg every 3 weeks for NSCLC previously treated with chemotherapy and for melanoma.72–74
Discussion
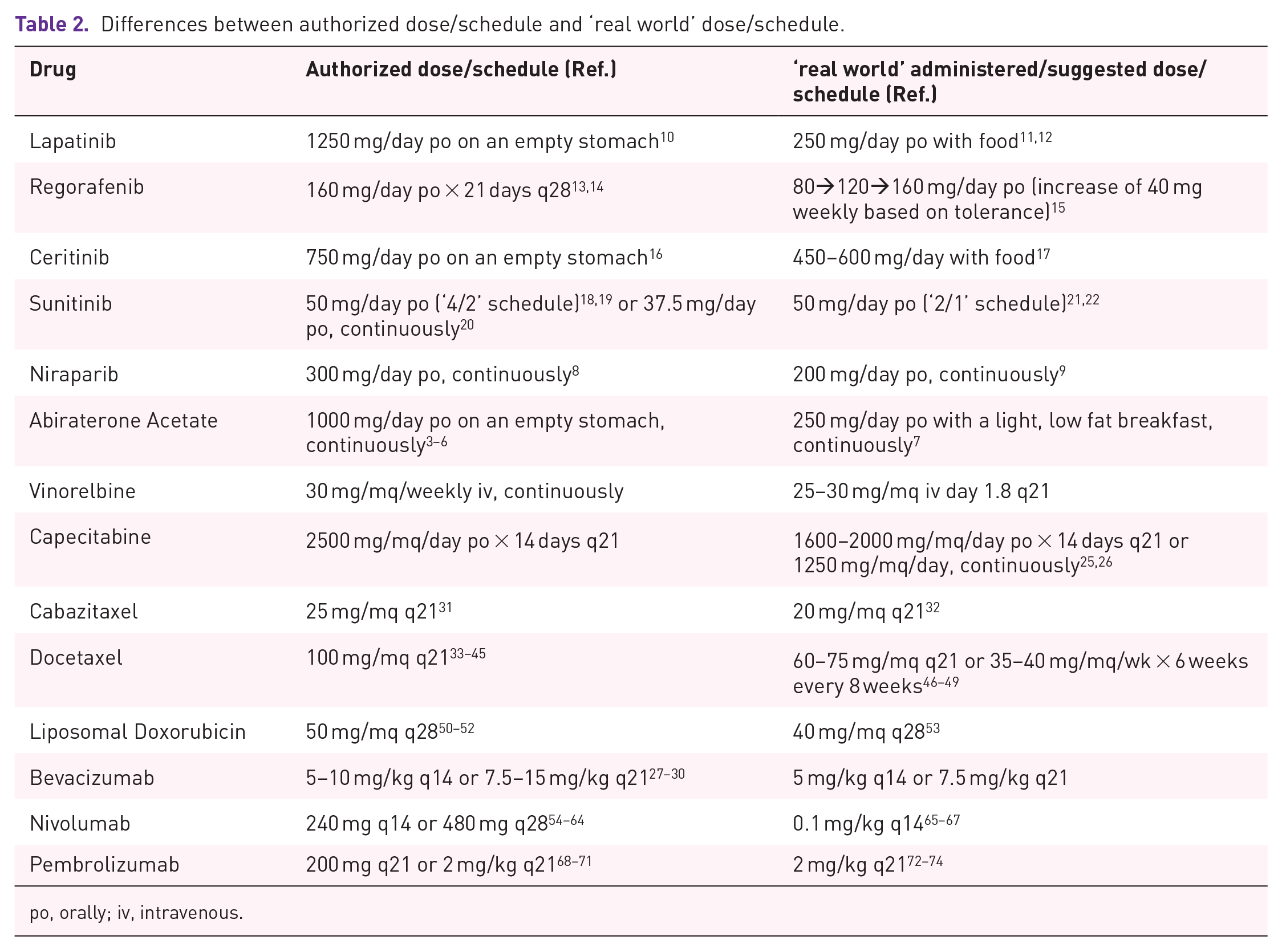
The history of medical oncology and haematology over the past 30 years has taught us that countless medicinal products, both chemotherapy agents and biologicals, are now used in clinical practice at different, usually lower, doses or with less intensive regimens than those for which they were authorized and marketed (Table 2). This happens for many reasons, some known, others more obscure, and lies primarily in the frequent biases present in the pivotal studies that often enrol patients who are not representative of the real population that is subsequently treated with that given medicinal product or that given regimen. It is no coincidence that an increasing number of postmarketing ‘real world’ observational studies are being conducted. For example, the enrolment in these studies of the elderly population >70 years of age is almost always lacking.
75
Otherwise the differences in the metabolism of the medicinal product between the different ethnic groups and different sexes are not considered
76
and the conclusions they reach regarding the toxicity observed at the maximum tolerated dose (MTD) are often reported in an ambiguous manner using unclear terminology; for example, ‘Most patients had an acceptable adverse-event profile’, or ‘. . .has a manageable and mostly reversible safety profile’, or ‘. . .the tolerability was good overall’ and ‘Incomplete reporting that downplayed drug-related adverse events was identified in 43% of reports of cancer drug trials’,
77
thereby generating the illusion of having identified the dose that is transversally best for all types of patient. Furthermore, a recent analysis showed that out of 101 pivotal studies on medicinal products approved by the FDA, a high incidence has been reported for even considerable changes once the study is under way to the sample size, the inclusion criteria and the primary endpoint. More specifically, 56 studies (equal to 55.4%) underwent a change in the sample size, 34 studies (equal to 33.7%) underwent changes to the inclusion criteria and 27 studies (equal to 26.7%) underwent a change in the primary endpoint. In the final publication, these changes were described in 39 (69.6%) out of the 56 cases with changes in the sample size, in 19 (55.9%) of the 34 cases with changes in the inclusion criteria, and in 10 out of the 27 studies (37.0%) in which the primary endpoint was modified.
78
‘Many drugs are now approved on the basis of whether they shrink the tumour or delay the time until the tumour grows, but they don’t necessarily help patients to live longer or better lives’. Consequently, it is also essential to assess the impact these medicinal products have on the quality of life. Therefore, the simple question we should be asking is: ‘Does the dose or regimen with which the medical product is marketed actually represent what is best for the patient in terms of efficacy and tolerability?’ The ‘old’ concept of MTD in phase I studies has most likely now been superseded and perhaps we should work on a new emerging concept, that of ‘minimum efficacious dose’, which is far closer to the real world setting. Indeed, in regards to targeted therapies, for example, the best way to determine the optimum dose is still unclear, although various alternative strategies to MTD have been explored. Most oncological agents have a strong dose–response correlation; minimal changes in the dose administered can lead to severe toxicity that can be life threatening in some patients, and to under dosing in others, such as to prejudice the result. Consequently, choosing the right dose is an aspect of great importance, especially in individuals with a potentially curable disease or in the adjuvant therapy setting. However, choosing the best dose is complicated by the fact that individuals have very varied abilities to metabolize and eliminate medicinal products. The most relevant pharmacokinetic parameter for exposure to a medicinal product is the area under the curve (AUC) of the plasma concentration × unit of time after the administration of a single dose. However, the AUC is influenced by both external factors, such as the dose of the drug or the regimen used, and by factors that are intrinsic to the patient, such as age, sex, weight, height and genetic factors such as the capacity to metabolize a drug or its clearance, which correlates closely with hepatic and renal function.
79
Therefore, in the attempt to minimize the potential subjective variables, the dose of most chemotherapy agents is still calculated on the basis of body surface area (BSA). The situation is completely different for targeted therapies, a very diverse and confusing field in which some medicinal products can be administered on the basis of body weight alone (e.g. monoclonal antibodies such as cetuximab, bevacizumab, intravenous trastuzumab, ipililumab, ramucirumab, panitumumab), others at a flat dose, such as a number of oral tyrosine kinase-inhibitors (e.g. dabrafenib, vemurafenib, trametinib, gefitinib, erlotinib, afatinib) or the m-TOR inhibitors, others still are administered both at doses calculated on the basis of body weight and flat doses such as certain immunotherapy agents (pembrolizumab, nivolumab, atezolizumab) or some monoclonal antibodies (e.g. alemtuzumab, ofatumumab, pertuzumab, subcutaneous trastuzumab). Finally, others, like carboplatin, are administered with doses calculated on the basis of the AUC.80,81 One paradigmatic example is olaparib, a PARP inhibitor, whose biological activity, considered as the inhibition of the enzyme in the tumour tissue, has been used to identify the most appropriate dose. In phase I studies, the biological activity of olaparib was demonstrated at doses achieved with two daily administrations of over 100 mg and with an MTD of 600 mg.82,83 The final dose subsequently approved by the FDA was 400 mg twice daily. Finally, despite the lack of robust data in favour of the routine use of BSA for dose calculation and an increasing number of scientific publications that question the validity of this method for a number of conventional cytotoxic agents,26,84–92 the dose of most chemotherapy drugs is still calculated on the basis of the BSA.93–96 Unfortunately, for most cancer drugs dose calculation using BSA has a limited ability to consider the individual variability in the clearance of the agent after the administration of a single dose.97,98 Oncologists are now aware that the administration of a drug calculated using BSA alone can have a subjective variability of over 30%,
81
whereas the dose calculated on the basis of clearance values can oscillate within a range of between 4- and 10-fold.99,100
po, orally; iv, intravenous.
In conclusion, the evidence available shows that we all too often see a posteriori changes in the doses of authorized regimens of a number of cancer drugs without there being any negative repercussions on costs; rather, there is often a gain in terms of tolerability and in cost savings. We are therefore of the opinion that we need to rethink phase I dose-finding studies based on the identification of the MTD and in the reporting of toxicity in general and serious adverse events in particular, by introducing the ‘minimum efficacious dose’ concept.