
Abstract
Multiomic analyses have shed light upon the molecular heterogeneity and complexity of triple-negative breast cancers (TNBCs). With increasing recognition that TNBC is not a single disease entity but encompasses different disease subtypes, a one-size-fits-all treatment paradigm has become obsolete. In this context, the inhibition of phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and androgen receptor (AR) signaling pathways have emerged as potential therapeutic strategies against selected tumors. In this paper, we reviewed the preclinical rationale, predictive biomarkers, efficacy, and safety data from early phase trials, and the future directions for these two biomarker-directed treatment approaches in TNBC.
Introduction
The past decade has witnessed concerted efforts to characterize triple-negative breast cancer (TNBC) geno-molecularly beyond its traditional immunohistochemical (IHC) definition.1–5 The discovery of candidate targetable oncogenic drivers have further promoted clinical trials of novel systemic treatments to address the unmet clinical needs of this biologically aggressive malignancy.
One year ago, we proposed a clinically pragmatic algorithm that categorized TNBC into five subgroups to guide rational treatment selection. We matched these subgroups, namely defective DNA repair, inflamed phenotype, androgen receptor (AR)-positive, PI3K/AKT/PTEN altered, and unique antigen-expressing, to systemic treatments including platinum and PARP inhibitors, immunotherapy, AR blockade, AKT inhibitors, and antibody–drug conjugates respectively. 6
The landmark results of IMpassion130 study7,8 heralded the arrival of immunotherapy as a treatment paradigm in TNBC. This also signposted the departure from times when the standard of care agents against TNBC were confined to cytotoxics and the median survival of metastatic disease was a dismal 11–14 months. The intention-to-treat (ITT) population in IMpassion130 attained a numerically longer median survival of 18.7 months 8 versus historical controls and highlights the stark shortfall in the prognosis of TNBC from HER-positive or luminal breast cancers.
We now recognize that TNBC is a heterogeneous disease, 9 and we are also starting to appreciate that early-stage breast cancers are genomically different from their metastatic counterparts. 10 For instance, among TNBC, the prevalence of somatic biallelic loss-of-function mutations in genes related to homologous recombination DNA repair is 3.5 fold higher in metastatic cases than in early cancers (7% versus 2%). Furthermore, metastatic breast cancers harbor greater mutational burden and clonal diversity compared with early cancers. 10 The genetic complexity of advanced breast cancers, including TNBC, is accompanied by an enrichment of clinically actionable genetic aberrations and offers valuable opportunities for molecularly rational therapeutic exploitation, even early in the disease course.
As we approach the end of this decade, we reviewed the two biomarker driven strategies of inhibiting the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and AR signaling pathways to treat TNBC in this paper.
PI3K/AKT inhibition
Preclinical rationale
The PI3K/AKT/mTOR signaling pathway is pivotal in carcinogenesis, promoting tumor survival, and growth.11,12 It is often activated in TNBC, and is not limited to the luminal androgen receptor (LAR) gene expression subgroup. 13
The high rate of PI3K/AKT/mTOR pathway aberrations is a distinctive finding of triple-negative, specifically basal-like, breast cancer in The Cancer Genome Atlas. Activation of the PI3K pathway is primarily mediated at the protein level and is less dependent on PIK3CA mutations (7%), but more commonly through the loss of negative regulators PTEN (mutation or loss, 35%) and INPP4B, or both (loss 30%). 3
Furthermore, deficient expression of PTEN is prevalent in TNBC and is associated with a greater degree of AKT pathway activation. 14
Ipatasertib is a highly selective oral ATP-competitive pan-AKT inhibitor which preferentially targets the phosphorylated conformation of AKT. 15 PI3K/AKT pathway activation is relevant for the survival of cancer cells under mitotic stress 16 and following exposure to chemotherapy. Activation of the PI3K/AKT pathway may confer resistance to taxanes. In contrast, in preclinical models, concurrent inhibition of the PI3K/AKT pathway enhances the efficacy of taxanes. Data from preclinical studies support the partnering of ipatasertib with paclitaxel for synergy. 17 Sensitivity to ipatasertib was associated with high phosphorylated AKT levels, PTEN protein loss, and mutations in PTEN or PIK3CA. 15
Similar to ipatasertib, capivasertib (AZD5363) is another highly selective, oral small molecular AKT inhibitor which binds to and inhibits all AKT isoforms. Capivasertib has shown preclinical activity in TNBC models with and without alterations of PIK3CA, AKT1 and PTEN, but sensitivity was associated with activation of PI3K or AKT, deletions of PTEN, or both. 18
Therapeutic inhibition of AKT, PI3K, and mTOR (mammalian target of rapamycin) triggers feedback loops which potentially limit the efficacy of these agents and can promote acquired resistance to single-agent receptor tyrosine kinase (RTK) inhibition. Specifically, AKT inhibition initiates FOXO-dependent transcription and activation of RTKs. 19 PI3K inhibition prevents downstream AKT activation but also induces enhanced mitogen-activated protein kinase (MAPK) signaling. 20 mTOR inhibition upregulates upstream RTKs, leading to the rebound activation of AKT. 21
Clinical drug development
Despite sound preclinical data, first-generation mTOR and PI3K inhibitors have produced mixed results in metastatic TNBC.22,23 This could be attributed to their lower target selectivity, uncontrolled activation of feedback loops when mTOR inhibitor was paired with chemotherapy, or toxicities limiting efficient drug delivery concomitant to chemotherapy. 24 Earlier trials commonly combined paclitaxel with an mTOR inhibitor,22,23 but there now is new preclinical evidence that phosphorylation of AKT could be activated by paclitaxel, a microtubule-stabilizing agent. In contrast, AKT phosphorylation was shown to be suppressed by eribulin, a microtubule-depolymerizing agent, in cell line and murine models. 25
Alpelisib is an oral, new generation, p100-alpha isoform-specific PI3K inhibitor. 26 PIK3CA-mutated cancers demonstrate sensitivity to alpelisib in preclinical tumor models 26 and in a phase I trial of alpelisib in patients with advanced solid tumors. 27 In a phase I/II study of alpelisib in combination with nab-paclitaxel to treat patients with metastatic HER2-negative breast cancer, 12 of the 42 enrolled subjects had a triple-negative disease. Median progression free survival (PFS) in the PIK3CA-mutated cohort was 13 months versus 7 months for the PIK3CA nonmutated cohort (HR 0.40, p = 0.017). 28
In contrast, AKT inhibitors which target an important node in the PI3K/AKT signaling cascade appear to be more promising in TNBC. In the adaptive phase II I-SPY2 trial, among tumors bearing the HR-/HER2- signature, the AKT inhibitor, MK-2206, plus standard neoadjuvant chemotherapy attained an estimated pathological complete response (pCR) rate of 40% compared with 22% from chemotherapy alone. 29
LOTUS is a randomized, double-blind, placebo-controlled, phase II study designed to investigate the efficacy of ipatasertib 400 mg on days 1–21 plus paclitaxel 80 mg/m2 on days 1, 8, and 15 every 28 days in treatment-naïve locally advanced or metastatic TNBC (n = 124).30,31 Subjects must have archival or newly obtained tumor tissue for central PTEN assessment. Tumor PTEN status defined by IHC (H-score 0 versus 1–150 versus >150) was a stratification factor. LOTUS met one of its two coprimary endpoints. PFS in the ITT population was modestly but significantly longer with ipatasertib versus placebo [6.2 months versus 4.9 months, the hazard ratio (HR) 0.60, p = 0.037]. However, the other coprimary endpoint was not reached: in the PTEN-low population (n = 48), ipatasertib did not significantly increase PFS (6.2 versus 3.7 months, HR 0.59, p = 0.18). 30 In an updated overall survival (OS) analysis, there was a trend toward better OS in the ITT population, with an almost 5 month difference in the medians between ipatasertib and placebo [23.1 versus 18.4 months, stratified HR 0.62 (95% confidence interval, 0.37–1.05)]. 31
Of note, treatment benefit derived from ipatasertib was greater in patients with PIK3CA/AKT1/PTEN altered tumors identified through next-generation sequencing. In prespecified analyses of this subgroup (n = 42), median PFS was 9.0 months with ipatasertib, and longer than 4.9 months with placebo (HR 0.44, p = 0.04). In contrast, in the 61 patients with PIK3CA/AKT1/PTEN nonaltered tumors, median PFS was 5.3 months versus 3.7 months in the ipatasertib and placebo groups respectively (HR 0.76, p = 0.36). 30 OS data stratified by biomarker status is immature. 31
The ipatasertib–paclitaxel doublet in LOTUS was generally tolerated. The most frequent of any grade adverse events (AEs) were gastrointestinal (diarrhea, nausea, and vomiting), alopecia, neuropathy, fatigue and rash, and typically graded 1 or 2 in severity. 30 Diarrhea was the most clinically relevant additive toxicity. Grade 3 or worse AEs were observed in 56% of the ipatasertib group compared with 44% in the placebo, 31 with diarrhea, neutropenia, peripheral neuropathy, fatigue and pneumonia being the most common. 30
LOTUS had been hypothesis generating and was followed by the further evaluation of frontline ipatasertib plus paclitaxel specifically for PIK3CA/AKT1/PTEN altered locally advanced or metastatic TNBC in the ongoing randomized phase III IPATunity130 trial (ClinicalTrials.gov identifier: NCT03337724).
PAKT is a randomized, double-blind, placebo-controlled, phase II trial which is analogous in design to LOTUS of first-line paclitaxel 90 mg/m2 on days 1, 8, and 15 with or without capivasertib 400 mg twice daily on days 2–5, 9–12 and 16–19 every 28 days (n = 140). 32 PFS by investigator assessment for the ITT population was the primary endpoint. Median PFS was significantly longer in the experimental arm compared with the placebo-controlled arm (5.9 versus 4.2 months, HR 0.74, one-sided p = 0.06). The secondary endpoint, OS, was also met (median OS 19.1 versus 12.6 months, HR 0.61, one-sided p = 0.02). Of note, efficacy was more pronounced in patients with PIK3CA/AKT1/PTEN altered tumors, adding capivasertib improved median PFS from 3.7 months to 9.3 months (HR 0.30, two-sided p = 0.01), but it did not result in PFS difference in the nonaltered group (5.3 versus 4.4 months, HR 1.13, two-sided p = 0.61). The most common AEs attributed to capivasertib were diarrhea, fatigue, rash and stomatitis. The incidence of grade 3/4 diarrhea was 13.2% in the capivasertib group compared with 1.4% in the placebo group. Unlike ipatasertib, capivasertib was associated with more cases of hyperglycemia.
AKT inhibition has also been studied in the neoadjuvant setting through the randomized phase II FAIRLANE trial (n = 151).33,34 Subjects with early TNBC (T ⩾ 1.5 cm, N0–2) were assigned 1:1 to receive weekly paclitaxel 80 mg/m2 with ipatasertib 400 mg or placebo on days 1–21 every 28 days for 12 weeks before surgery. Coprimary endpoints were pCR rate (ypT0/TisN0) in the ITT and IHC PTEN-low populations. Secondary endpoints included the pCR rate in patients with PIK3CA/AKT1/PTEN altered tumors and pre-surgery response rates by magnetic resonance imaging (MRI). The addition of ipatasertib to neoadjuvant paclitaxel did not clinically, or statistically, significantly increase the pCR rate, although the overall response rate (ORR) by MRI was numerically higher with ipatasertib. The antitumor effect of ipatasertib was most pronounced in biomarker-selected patients. All patients with a complete response had PIK3CA/AKT1/PTEN altered tumors. 33
The rationale for combination with immunotherapy
Loss of PTEN, a negative regulator of AKT, has been found to be a potential mechanism of resistance to immune checkpoint blockade. Antagonizing the PI3K/AKT pathway with a PI3Kβ inhibitor was shown to reverse resistance to T cell-mediated immunotherapy. 35 AKT inhibitors may restore or augment the physiological function of T cells in the tumor microenvironment, and promote the expansion of tumor-specific lymphocytes with stem-like memory cell phenotype.36,37 Co-administration of ipatasertib may increase checkpoint inhibitor efficacy by retaining a stem-like phenotype in memory T cells, preventing exhaustion and enabling a long-term response in patients.38,39 Dual PI3Kβ inhibition and programmed death-ligand 1 (PD-L1)/programmed death-1 (PD-1) axis blockade elicited synergistic anticancer responses. 35
Future directions
Preliminary results from CO40151, a multicenter phase IB study evaluating a triplet of ipatasertib, atezolizumab, and paclitaxel or nab-paclitaxel for unresectable locally advanced or metastatic TNBC revealed an unprecedented 73% ORR, independent of biomarker status. The most commonly reported AEs were diarrhea and rash which were manageable. 40
This proof of concept study provided the basis for the Cohort C of IPATunity130 trial in a recent protocol amendment at selected sites which will explore the triplet of ipatasertib, paclitaxel, and atezolizumab in TNBC patients with PI3K/AKT/PTEN nonaltered tumors. A similar triplet approach was also evaluated in one of the experimental arms of the ongoing phase IB/II BEGONIA trial (ClinicalTrials.gov identifier: NCT03742102) which features capivasertib, paclitaxel, and durvalumab.
Other trials of PI3K/AKT/mTOR inhibitors in conjunction with immune checkpoint blockade and other partners are presented in Table 1.
Active and completed trials of PI3K/AKT/mTOR inhibition in locally advanced and metastatic triple negative breast cancer.
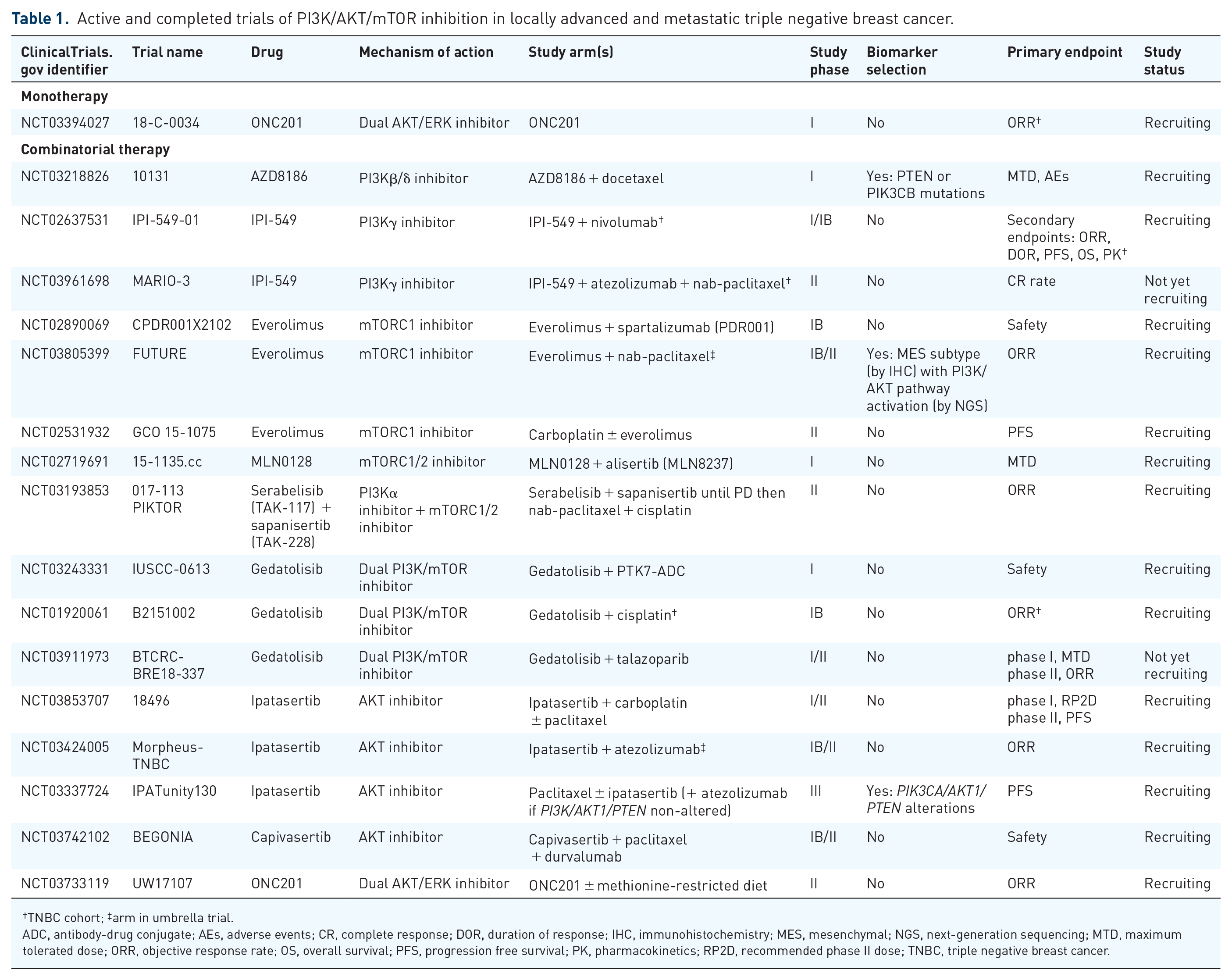
TNBC cohort; ‡arm in umbrella trial.
ADC, antibody-drug conjugate; AEs, adverse events; CR, complete response; DOR, duration of response; IHC, immunohistochemistry; MES, mesenchymal; NGS, next-generation sequencing; MTD, maximum tolerated dose; ORR, objective response rate; OS, overall survival; PFS, progression free survival; PK, pharmacokinetics; RP2D, recommended phase II dose; TNBC, triple negative breast cancer.
Dual inhibitors of the PI3K/AKT/mTOR pathway may control target pathway activation and the resulting feedback loops, thus circumventing treatment resistance. In patient-derived xenograph models of basal-like breast cancer, combined antagonism of mTOR and AKT was synergistic. 41 The clinical development of dual inhibitors is impaired by unacceptable toxicities although several remain under investigation, including gedatolisib.
In breast cancers, crosstalk can exist at multiple levels between the pro-survival PI3K/AKT and the mitogenic RAS/RAS/MEK/ERK (also known as MAPK) pathways. 42 Extracellular signal-regulated kinase (ERK) and mitogen-activated protein kinase kinase (MEK) are major cassettes of the MAPK signaling cascade.
ONC201 is a first-in-class imipridone which is being studied in multiple cancers including TNBC. It was initially reported to inhibit AKT and ERK phosphorylation, leading to dephosphorylation of FOXO3a, and the transcriptional induction of the pro-apoptotic protein tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL). 43 However, this mechanism of action has been questioned because, in breast and endometrial cancer cell lines, the cytotoxicity of ONC201 appeared to be mediated through disruption of the mitochondrial function instead. 44
Phase I and II trials exploring the safety and preliminary efficacy of gedatolisib and ONC201, mainly as combination treatments, in advanced TNBC are also listed in Table 1.
More recently, a preclinical study demonstrated that the dual inhibition of PI3K/AKT and MEK5/ERK5 pathways using ipatasertib and a novel MEK5 inhibitor (SC-1-181), respectively, on TNBC cell lines resulted in deleterious effects on cell viability and survival, to a greater degree than inhibition of either pathway alone. This synergy was apparently mediated through the loss of BCL2 associated agonist of cell death (BAD) phosphorylation, restoration of p21, decreased cell proliferation, and enhanced apoptosis. 45
Preclinical research also identified the Receptor Orphan Tyrosine Kinase-like Receptor-1 (ROR1) pathway as another signaling network which intersects with the PI3K/AKT pathway in TNBC. ROR1 signaling culminates in AKT activation via phosphorylation of serine 473 by PI3K. ROR1 antagonism, therefore, arrested proliferation in TNBC. In TNBC cell lines, ROR1 inhibition with a novel antagonist, strictinin, reduced AKT phosphorylation on serine 473, inhibiting downstream phosphorylation of glycogen synthase kinase 3 beta (GSK3β). Through the reactivation of GSK3β, strictinin also impaired cell migration and invasion in TNBC. 46
Further preclinical work needs to be carried out to validate the in vitro and in vivo effects of dual MEK-AKT inhibition or ROR1 antagonism in TNBC before the clinical development of either therapeutic strategy can be launched.
AR blockade
Preclinical rationale
The LAR subtype of TNBC is one of the four intrinsic molecular subtypes in Lehmann’s revised taxonomy based on gene expression profiling, comprising 16% by proportion. 1 In contrast, using Perou’s schema, 20–30% of TNBCs are stratified as luminal/AR. 2 LAR TNBCs show high transcription of AR messenger RNA in addition to downstream AR targets and coactivators. 47 There is significant variability reported in the incidence (7–75%) and prognostic significance of AR expression in TNBC. 48 Clinical and preclinical research suggests that AR acts as a tumor suppressor in estrogen receptor (ER)-positive breast cancers and as an oncogene product in ER-negative cases. 49 Preclinical in vitro47,50 and in vivo51,52 data further suggest that androgens mediate the development and growth of the MDA-MB-452 cell line which serves as an in vitro model for AR-positive TNBC. Therefore, targeting AR, similar to hormone-sensitive prostate cancer, has been pursued as a therapeutic strategy in AR-positive TNBC patients.
Immunohistochemical staining employing polyclonal antibodies to the ligand-binding domain is widely used to define AR positivity in breast cancer. 53 It is not known what cutoff value should be used to predict response to AR inhibition in breast cancer, as evident from the clinical studies showcased below and in Table 2.
Active and completed trials of androgen blockade in triple-negative breast cancer.

TNBC cohort; ‡in primary or metastatic lesion.
AEs, adverse events; AR, androgen receptor; CBR, clinical benefit rate; DCR, disease control rate; DLT, dose-limiting toxicities; GC, gemcitabine-carboplatin; GT, gemcitabine-paclitaxel; IHC, immunohistochemistry; MTD, maximum tolerated dose; ORR, objective response rate; pCR, pathologic complete response; PFS, progression free survival; RCB-I, residual cancer burden-index; RP2D, recommended phase II dose; SARM, selective androgen receptor modulator; TNBC, triple-negative breast cancer; TPC, treatment of physician’s choice; TX, docetaxel-capecitabine.
Clinical drug development
The drug classes evaluated in contemporary trials of AR pathway inhibition in TNBC are nonsteroidal selective AR antagonists, CYP17A inhibitors, 7-hydroxytestosterone, and selective androgen receptor modulators (SARMs).
The first of early phase AR-focused trials in advanced TNBC was the TBCRC011 study which assessed bicalutamide, an AR antagonist, among 51 hormone receptor-negative patients screened positive for AR (>10% by IHC). A 24-week clinical benefit rate (CBR) of 19% was observed, with no overall responses, median PFS was 12 weeks. 54 This proof of principle study was followed by other phase II single-arm trials.
The phase II UCBG 12-1 trial of abiraterone acetate, a CYP17A inhibitor, in a cohort of heavily pretreated AR-positive (⩾10% by IHC) TNBC showed a 6-month CBR of 20% and a PFS of 2.8 months. 55
Similarly, in a phase II MDV3100-11 trial of enzalutamide, a potent AR inhibitor, the 16-week CBR was 33%, and a median PFS of 3.3 weeks was seen among the 78 evaluated AR-positive advanced TNBC patients. 56 In MDV3100-11, AR positivity was defined as >0% by IHC. The exploratory analysis also found that an androgen-related gene signature obtained from a genomic diagnostic assay, PREDICT AR, was associated with a greater clinical benefit.57,58
The phase III three-arm ENDEAR trial (ClinicalTrials.gov identifier: NCT02929576) of paclitaxel plus enzalutamide versus placebo or enzalutamide monotherapy followed by paclitaxel was designed in diagnostic signature-positive advanced TNBC. However, ENDEAR was withdrawn, pending further understanding about the role of AR signaling in TNBC.
Future directions
Newer agents under development for AR-positive metastatic breast cancer (either TNBC or hormone receptor-positive) include darolutamide an AR antagonist, orteronel (TAK-700) nonsteroidal CYP17A1 inhibitor, seviteronel (VT-464) a dual CYP17 inhibitor-cum-AR antagonist, and enobosarm (ostarine, GTx-024), a SARM. A phase II study of transdermal enobosarm monotherapy in patients with AR-positive TNBC (ClinicalTrials.gov identifier: NCT02368691) was terminated owing to a lack of efficacy.
Combination approaches pairing AR blockade with small molecule inhibitors, chemotherapy, or immunotherapy are under investigation in early and advanced disease settings. Active and completed clinical studies incorporating AR blockade in the treatment of TNBC are summarized in Table 2.
AR-positive TNBCs are enriched for PIK3CA mutations compared with AR-negative TNBCs.14,59 Referring back to the other therapeutic direction described earlier in this review, adding PI3K/AKT inhibition to AR antagonism has shown synergistic activity in AR-positive TNBC preclinical models. A phase I/II trial is recruiting patients with triple-negative and hormone receptor-positive tumors which are positive for AR and PTEN by IHC, and examining the efficacy and safety of alpelisib plus enzalutamide (ClinicalTrials.gov identifier: NCT03207529). A similar combination of another PI3Kα inhibitor, taselisib, plus enzalutamide (ClinicalTrials.gov identifier: NCT02457910) is also being studied. However, it is worth noting that regulatory approval was not sought for taselisib plus fulvestrant in patients with ER-positive, PIK3CA-mutant advanced breast cancer after the phase III SANDPIPER trial reported a modest improvement in PFS at the expense of a challenging safety profile which is attributable to taselisib. 60
Unresolved issues
An unresolved question pertaining to the AR-positive subgroup of TNBC is how best to define AR positivity. The status quo is that there is no universal consensus of AR positivity, unlike its counterparts of estrogen, progesterone, and HER2 receptors. As mentioned earlier, all concluded and ongoing clinical studies have employed IHC, but at different threshold values, to select patients for AR inhibition whereas the LAR subtype of TNBC is conventionally defined by gene expression profiling. The androgen-related gene signature which was studied as an exploratory objective in MDV3100-11 requires validation in other datasets before gaining widespread acceptance as being predictive of anti-AR response. Harmonizing a rational strategic approach to identify the subgroup of TNBC patients who will benefit from AR blockade remains an unmet need. Without this, the pursuit of phase III trials such as ENDEAR will continue to be impeded.
Another unresolved issue is the question of de-escalation versus the escalation of systemic treatments. We do not yet, to the best of our knowledge, have criteria to stratify AR-positive TNBC patients to those who are well served by monotherapy with an excellent tolerability profile versus those who require combined treatment with the vertical integration of other drugs in a scientifically based manner. Indeed, in Table 2, we continue to see trials of androgen blockade alone as a single-arm in pre-operative cases, or in comparison with cytotoxic chemotherapy in the advanced stages. This sort of study design reflects the common clinical observation that a significant proportion of AR-positive tumors follow a less aggressive course and fare prognostically better than other subtypes of TNBCs.
Discussion
We have provided insights into two emerging but disparate methods for treating TNBC and demonstrated some ongoing work to even amalgamate both approaches. In the reported and ongoing trials involving AKT inhibitors, concurrent taxane chemotherapy has been utilized to target synergistic outcomes. Conversely, AR blockade by itself seems to have limited efficacy with few objective responses as a chemotherapy-free alternative in TNBC.
To date, PI3K/AKT inhibition is enjoying a greater measure of success in the drug development process than the androgen blockade. In this regard, the results of IPATunity130 are eagerly awaited. It will be interesting to see whether the impressive result of triplet therapy of AKT inhibitor, taxane and anti-PD-L1 antibody studied in CO40151 can be replicated in Cohort C of IPATunity130.
However, a breakthrough is required for AR blockade to advance from early phase investigation to phase III trials. AR inhibition continues to be hampered by mechanistic constraints and imprecise patient selection because of issues relating to assay methodology and cutoff for biomarker positivity.
As we look forward to the next decade, the treatment of TNBC may continue to be supported by cytotoxic chemotherapy. We will rely on robust biomarkers and positive, well-designed, prospective, randomized trials to make the prospect of biomarker-selected therapeutics a reality in the near future. Newer technologies to elucidate the geno-molecular underpinnings of this complex disease may also translate to treatment opportunities.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflicts of interest statement
Jack J Chan: Honoraria; Merck Sharp & Dohme, Eisai, AstraZeneca, Novartis; Consulting or Advisory Role, Pfizer; Research Funding, OncoQuest; Travel, Accommodations, Expenses, AstraZeneca, Celgene.
Tira JY Tan: Consulting or Advisory Role; Roche, Novartis, Eli Lily; Research Funding; AstraZeneca; Travel, Accommodations, Expenses, Eisai.
Rebecca A Dent: Honoraria; AstraZeneca, Lilly, Pfizer, Roche/Genentech; Consulting or Advisory Role; AstraZeneca, Eisai, Merck, Novartis, Pfizer, Roche; Travel, Accommodations, Expenses; Amgen, Merck, Pfizer, Roche.