
Abstract
Wnt/β-catenin and Hippo pathways play essential roles in the tumorigenesis and development of colorectal cancer. We found that Celastrol, isolated from Tripterygium wilfordii plant, exerted a significant inhibitory effect on colorectal cancer cell growth in vitro and in vivo, and further unraveled the molecular mechanisms. Celastrol induced β-catenin degradation through phosphorylation of Yes-associated protein (YAP), a major downstream effector of Hippo pathway, and also Celastrol-induced β-catenin degradation was dependent on liver kinase B1 (LKB1). Celastrol increased the transcriptional activation of LKB1, partially through the heat shock factor 1 (HSF1). Moreover, LKB1 activated AMP-activated protein kinase α (AMPKα) and further phosphorylated YAP, which eventually promoted the degradation of β-catenin. In addition, LKB1 deficiency promoted colorectal cancer cell growth and attenuated the inhibitory effect of Celastrol on colorectal cancer growth both in vitro and in vivo. Taken together, Celastrol inhibited colorectal cancer cell growth by promoting β-catenin degradation via the HSF1–LKB1–AMPKα–YAP pathway. These results suggested that Celastrol may potentially serve as a future drug for colorectal cancer treatment.
Introduction
As the most common gastrointestinal (GI) tract cancer worldwide, colorectal cancer (CRC) represents the fifth leading causes of cancer deaths among both men and women. 1 CRC acquires many genetic alterations, and some signaling pathways involved are clearly singled out as key factors in tumor formation. Activation of the Wnt pathway is regarded as the initiating event in CRC. 2 The core of Wnt signaling is β-catenin, which is regulated by a cytoplasmic destruction complex consisting of a central scaffold protein Axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK-3β), and casein kinase 1 (CK1). 3 As an critical transcriptional co-activator of the Wnt pathway, β-catenin regulates target gene expression. 4 Aberrant expression of β-catenin induces malignant transformation of normal cells, and its abnormal activity has been reported in many cancer types.
Hippo signaling interacts with Wnt/β-catenin pathway. 5 Activation of the Hippo pathway leads to phosphorylation of Yes-Associated Protein (YAP) and transcriptional coactivator with PDZ binding motif (TAZ) [also referred to as WW-domain containing transcriptional regulator 1 (WWTR1)]. 6 Accumulating evidence has strongly suggested the critical role of dysregulated YAP in the tumorigenesis. YAP is required for the development of APC-deficient adenomas. 7 Hyperactivated YAP results in widespread early onset polyp formation following dextran sulfate sodium (DSS) treatment that induces inflammatory bowel disease. 8 In addition, YAP promotes the nuclear accumulation of β-catenin in intestinal regeneration and the survival of β-catenin-driven colon cancers.9,10 Transgenic expression of YAP reduces Wnt target gene expression and results in the rapid loss of intestinal crypts. 11 Previous studies showed that APC (a negative regulator of β-catenin) could dually regulate YAP and β-catenin through parallel pathways involving the Hippo kinase cascade and the β-catenin destruction complex, respectively.5,7 These observations suggest the important interactions of β-catenin and YAP in CRC.
LKB1/AMPKα pathway is a ubiquitous pathway that controls a wide range of cellular functions including metabolism, proliferation, and cell shape. 12 LKB1 inactivation is frequently observed in a variety of cancers.13,14 AMPKα, a LKB1 main downstream target, is an intracellular energy sensor involved in cancer progression. 15 Recent reports showed cross-talk of LKB1/AMPKα pathway and Hippo pathway in the central nervous system, 12 and LKB1-deficient cancers exhibit reduced Hippo kinase activity and enhanced YAP-driven transcription.16,17 Furthermore, LKB1 inhibits cell proliferation by suppressing the nuclear translocation of YAP and β-catenin in gastric cancer cells. 18 All these prompted that LKB1/ AMPKα pathway and Hippo pathway had inseparable relationships in human cancers.
Celastrol, a triterpene, is a pharmacologically active ingredient initially isolated from the roots of the Tripterygium wilfordii plant, which has anti-inflammatory, immune suppression, and antitumor activity.19,20 Celastrol induces cell cycle arrest and apoptosis,21,22 and acts as inhibitors of heat shock protein 90 (HSP90), 23 nuclear factor (NF)-κB, 24 and proteasome. 25 Celastrol activates HSF1 and heat shock response (HSR).26,27 HSR is a highly conserved ancient process that helps to maintain protein homeostasis and is essential for cell survival. 27 In response to stress, activated HSF1 exerts pleiotropic effects. 28 Celastrol ameliorates DSS-induced colitis and ulcerative colitis-related CRC in mice via modulating oxidative stress, inflammatory responses, epithelial–mesenchymal transition and intestinal homeostasis.29,30 The earliest study showed that β-catenin mediated the apoptosis induction effect of Celastrol in HT29 cells. 31 Then, Lin et al. indicated that Celastrol treatment significantly prevented azoxymethane (AOM)/DSS-induced upregulation of β-catenin expression. 30 However, the mechanisms were not well understood.
In the present study, we investigated molecular mechanisms of Celastrol in β-catenin regulation through activating the LKB1–AMPKα pathway and phosphorylating YAP. We also treated C57BL/6J-ApcMin/+ (APCMin/+) and AOM-DSS mouse models with Celastrol, and found that Celastrol exerted a significant inhibitory effect on CRC growth in vivo. Our findings indicate the molecular mechanisms underlying Celastrol regulation on β-catenin expression, and shed light on potential application of Celastrol in CRC treatment.
Materials and methods
Cell line, cell culture, and transfections
HEK293, HCT116, and SW480 cell lines were all purchased from the Cell Resource Center (Beijing, China) and identified by short tandem repeat (STR) analysis.T-Rex-293 cell line was presented by Professor Quan Chen (Chinese Academy of Sciences, Beijing, China).The inducible expression system of β-catenin cell line named T-Rex-293/β-catenin (S37A) was established as described previously. 32 HCT116 and SW480 cells were cultured in RPMI 1640 medium (BIOROC) containing 10% fetal bovine serum in a humidified environment at 37°C with 5% CO2. Cells were transfected in 70–80% confluency using Attractene (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Stably transfected cells were selected with 10 μg/ml puromycin for 14 days.
Reagents and plasmids
Celastrol (purity > 98%) was purchased from Mingrui Inc. (Shanghai, China). Cycloheximide (C4859) was purchased from Sigma-Aldrich (St. Louis, MO, USA). PS-341 (Bortezomib, No. S1013) and Compound C (Dorsomorphin 2HCl, NO.S7306) were purchased from Selleck Chemicals (USA). MDL-28170 and HSF1 siRNAs (sc-35611) were purchased from Santa Cruz (USA). YAP siRNA was purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA). HSF1 expression plasmid (CH864887) was purchased from Vigene Biosciences (Shandong, China). YAP and YAPS127A expression plasmids were constructed as described previously. 33 A DNA fragment covering the LKB1 coding region was synthesized and constructed into pcDNA4/TO/myc-His B (Invitrogen, Carlsbad, CA, USA).
MTT assay
A total of 3000 HCT116 and SW480 cells were seeded into 96-well culture plate per well, and treated with Celastrol (0.75 μM) for 5 days. Nontreated cells were used as controls and noncell wells with medium used as blank. MTT assay was performed as described previously. 34
Cell colony formation assay
A total of 800 cells were plated to 6-well plates per well and treated with Celastrol (0.75 μM). Medium with or without Celastrol (0.75 μM) was changed every 4 days. After 2 weeks, cells were washed with phosphate-buffered saline (PBS), fixed with formaldehyde, and stained with Giemsa staining solution. Nontreated cells were used as controls. Clones larger than 100 μm in diameter were counted under an Olympus inverted microscope.
Western blot
Cells were harvested and lysed in RIPA buffer (Cell Signaling Technology). Nuclear and cytoplasmic proteins were separated by NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, No. 78833). Western blot analysis was performed with the use of conventional protocols as described previously. 35 Primary antibodies used were: β-actin (1:5000, Sigma-Aldrich, A1978), PARP (1:1000, Cell Signaling Technology, No. 9542), YAP (1:1000, Cell Signaling Technology, No. 4912), pho-YAP(Ser127) (1:1000, Cell Signaling Technology, No. 13008), pho-β-catenin (Ser33/37/Thr41) (1:1000, Cell Signaling Technology, No. 9561), pho-GSK-3β (Ser9) (1:1000, Cell Signaling Technology, No. 9336), HSF1 (1:1000, Cell Signaling Technology, No. 12972), Axin1 (1:1000, Cell Signaling Technology, No. 2087), AMPKα (1:1000, Cell Signaling Technology, No. 5831), pho-AMPKα (Thr172) (1:1000, Cell Signaling Technology, No. 2535), Ubiquitin (1:1000, Cell Signaling Technology, No. 3936), β-Trcp (1:1000, Cell Signaling Technology, No. 4394), β-catenin (1:1000, Santa Cruz, sc-7963), LKB1 (1:1000, Santa Cruz, sc-32245), Lamin B (1:1000, Santa Cruz, sc-6217), c-myc (1:1000, Santa Cruz, sc-789), and Survivin (1:1000, Santa Cruz, sc-10811). Each Western blot was repeated at least three times for reproducibility and the intensities of the bands were analyzed using ImageJ software.
Quantitative real-time polymerase chain reaction (Q-PCR)
Quantitative real-time polymerase chain reaction (Q-PCR) was performed as described previously. 34 All the primers used are listed in Supplementary Table 1.
Luciferase reporter assay
TOPFLASH or FOPFLASH reporter and control plasmid pRL-TK were transfected into HEK293 cells, respectively. After transfection for 24 h, cells were treated with or without Celastrol (0.5 μM) for another 24 h. Luciferase activity was detected using the Dual Luciferase Reporter system (Promega) according to the manufacturer’s instructions.
Immunofluorescence
Cells or frozen sections at 5 μm thickness were fixed in 4% paraformaldehyde and methanol for 20 min at room temperature, respectively. Immunofluorescence was performed as described previously. 33 Antibodies used were including anti-β-catenin (1:1000, BD, 610154), anti-YAP (1:500, Cell Signaling Technology, NO.14074), Dylight 649-goat anti-mouse IgG (1:500, Earth Ox Life Sciences, E032610-01), and Dylight 488-goat anti-rabbit IgG (1:500, EarthOx Life Sciences, E032220-01).
TdT-mediated dUTP nick end labeling assay
Sections at 5 μM thickness were confirmed with in situ cell death detection kit, alkaline phosphatase (Roche Applied Science), in accordance with the manufacturer’s instructions. Apoptotic cells (dark blue staining) were counted three times in five random fields of vision under microscope.
Immunoprecipitation assay
Six million cells were lysed using nondenaturing lysis buffer (Pu Lilai Gene Technology, Beijing, China). The cell lysates were incubated with 30 μl of nonimmune IgG protein G-Sepharose for 1 h at 4°C and then centrifuged. The supernatant was incubated sequentially with 1–2 μg of appropriated antibodies and 30 μl of protein G-Sepharose (IgG as control) overnight at 4°C, and washed with nondenaturing lysis buffer for 3 times. Precipitated proteins were identified by Western blot analysis.
Chromatin immunoprecipitation assay
Eight million HCT116 cells were cross-linked and lysed. Chromatin immunoprecipitation (ChIP) assay was performed as described previously. 33 The primers that used to detect the LKB1 promoter region are listed in Supplementary Table 2. ChIP assay was repeated at least three times and representative results are shown.
Immunohistochemistry
Immunohistochemistry was performed as described previously. 36 Primary antibodies were used including anti-β-catenin (1:1000, BD, 610154), anti-HSF1 (1:200, Cell Signaling Technology, No. 12972), anti-LKB1 (1:100, Santa Cruz, sc-32245), and anti-PCNA (1:1000, Santa Cruz, sc-7907).
Tumor xenograft model
Two million cells were suspended in 0.1 ml of saline and injected subcutaneously into 6-week-old female Balb/c nude mice. Four different groups of cells were injected including HCT116, HCT116/LKB1-KO, SW480, and SW480/LKB1-KO, and each group consisted of five animals. Three weeks after tumor cell injection, mice were intraperitoneally injected with Celastrol for 2 weeks (1 mg/kg, three times a week), and sacrificed 1 week after the last Celastrol injection. Tumor xenografts were separated and measured at the end of the experiment.
In vivo experiments
C57BL/6J-APCMin/+ male mice were purchased from Jackson Laboratory (Bar Harbor, ME) and then bred with wildtype C57BL/6J female mice to obtain APCMin/+ alleles that were identified by PCR assays as described previously. 37 Twelve-week-old APCMin/+ mice were intraperitoneally injected with Celastrol (1 mg/kg) or PBS as control for 3 weeks (three times/week). Mice were sacrificed at 18 weeks old after treatment.
The AOM/DSS-induced mouse model was established as described previously. 38 At the second cycle, mice were intraperitoneally injected with Celastrol (1 mg/kg) or PBS for 3 weeks (three times/week), and sacrificed at 18 weeks old.
LKB1loxp/loxp mice were purchased from Jackson Laboratory (Bar Harbor, ME). Pvillin-Cre mice were purchased from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). LKB1loxp/loxp mice were crossbred with Pvillin-Cre mice to generate intestinal-specific LKB1-KO mice. Genotypes were determined by PCR using genomic DNA extracted from tails. Both heterozygous deletion (Cre+ LKB1+/loxp) and homozygous deletion (Cre+ LKB1loxp/loxp) of LKB1 mice were treated with AOM/DSS and received intraperitoneal injection of Celastrol or PBS as described previously. Mice were sacrificed at 18 weeks old.
All animals were handled in strict accordance with good animal practice as defined by the Beijing Municipal Science and Technology Commission, and all protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of National Cancer Center/Cancer Hospital (Approval No. NCC2015A019).
Statistical analysis
Results were presented as mean ± standard deviation (SD) or ± standard error of the mean (SEM). Student’s two-tailed nonpaired t-test was used to determine significance between treatment and control groups in all experiments. p < 0.05 was considered statistically significant.
Results
Celastrol inhibited cell growth and induced cell apoptosis in CRC
To investigate the effect of Celastrol on CRC cell growth, MTT and colony formation assays were performed in SW480 and HCT116 cells. Celastrol significantly inhibited cell growth (up to 60.14% reduction in SW480 and 46.61% reduction in HCT116 cells) and colony formation (up to 64.13% reduction in SW480 and 99.49% reduction in HCT116 cells) at a concentration of 0.75 μM as compared with nontreated control cells (Figure 1A and B and Supplementary Figure S1A). Furthermore, we used C57BL/6J-ApcMin/+ (APCMin/+) and AOM/DSS-treated mice, and intraperitoneally injected Celastrol for 3 weeks (1 mg/kg, 3 times/week) to assess the suppression effect of Celastrol on colorectal tumorigenesis in vivo. As shown in Figure 1C and D, in contrast to nontreated mice, Celastrol injection significantly reduced the tumor numbers from 38.6 ± 4.1 (n = 15) to 13.1 ± 2.2 (n = 18) (mean ± SEM, p < 0.0001) in the small intestine of APCMin/+ mice, and decreased the tumor numbers from 12.8 ± 2.3 (n = 5) to 4.3 ± 1.0 (n = 7) (mean ± SEM, p < 0.01) in the colon of AOM/DSS mice. Notably, Celastrol treatment showed an equivalent efficacy to 5-Fu, which is widely used for CRC treatment (Supplementary Figure S1B and C). Moreover, immunohistochemistry staining results showed that Celastrol treatment decreased proliferating cell nuclear antigen (PCNA) expression in the small intestine of APCMin/+ mice (17.1% in Celastrol versus 95.2% in controls) and in the colon of AOM/DSS mice (27.6% in Celastrol versus 93.0% in controls), as compared with PCNA expression in control mice (Figure 1E and F). In addition to inhibiting proliferation marker PCNA expression, we also detected Celastrol-induced cancer cell apoptosis. Celastrol treatment at 0.75 μM for 24 h enhanced PARP cleavage in SW480 and HCT116 cells (Figure 1G) in vitro, and also increased apoptotic cells in intestinal tumors in APCMin/+ mice (93.4% in Celastrol versus 21.9% in control) and in colorectal tumors in AOM/DSS mice (90.0% in Celastrol versus 19.1% in controls) as assessed by TdT-mediated dUTP nick end labeling (TUNEL) assay (Figure 1H and I). These data indicated that Celastrol inhibited intestinal cancer/CRC cell growth by inhibiting proliferation and inducing apoptosis.

Celastrol inhibited cell growth and induced cell apoptosis in colorectal cancer.
Celastrol induced β-catenin degradation through the ubiquitin–proteasome pathway
Dysregulated APC/β-catenin signaling pathway is an early and common event in CRC, therefore we examined whether Celastrol affects APC/β-catenin signaling. Western blot results showed that β-catenin abundance were reduced by Celastrol treatment in a dose- and time-dependent manner in both SW480 and HCT116 cells (Figure 2A and Supplementary Figure S2A). Celastrol also significantly decreased β-catenin transcriptional activity (74.2% reduction) as assessed by TOP/FOP luciferase assay (Supplementary Figure S2B). In addition, Celastrol decreased both mRNA and protein levels of β-catenin downstream genes including c-Myc, survivin, CYR61, and Cyclin D1 in SW480 and HCT116 cells (Supplementary Figure S2C and D). Similar results were also observed in mice models. Celastrol treatment reduced β-catenin protein levels in the intestinal mucosa of APCMin/+ mice and the colon of AOM/DSS mice (Figure 2B) as measured by Western blot. Immunohisto-chemistry staining of β-catenin also showed significantly suppression by Celastrol in these mouse tissues (Figure 2C). Furthermore, Celastrol suppressed the mRNA levels of survivin and c-Jun in intestinal or colorectal tumor tissues of APCMin/+ and AOM/DSS mice (Supplementary Figure S2E). Interestingly, expression of constitutively activated β-catenin (β-catenin S37A) rescued the Celastrol-induced PARP cleavage and also promoted cell colony growth as compared with the Celastrol-treated cells without β-catenin S37A transfection, which suggested that sustained activation of β-catenin could attenuate Celastrol-induced cell apoptosis (Supplementary Figure S3A and B).

Celastrol induced β-catenin degradation through the ubiquitin–proteasome pathway.
Different from the change of protein expression, β-catenin mRNA levels were not altered by Celastrol treatment as assessed by Q-PCR, suggesting that β-catenin protein reduction was not due to transcriptional alteration (data not shown). Therefore, we studied Celastrol involvement in β-catenin degradation. SW480 and HCT116 cells were treated with protein synthesis inhibitor cycloheximide (CHX), accompanied with or without Celastrol treatment at 0.75 μM. CHX treatment alone did not significantly affect β-catenin degradation up to 24 h. However, co-incubation of CHX and Celastrol for 12 hours started to significantly decrease β-catenin protein levels in HCT116 and SW480 cells (Figure 2D). These results implied Celastrol involvement in β-catenin protein degradation. Thus we utilized MDL-28170 (a calpain inhibitor III) and PS-341 (a classic proteasome inhibitor) to pre-treat SW480 and HCT116 cells prior to Celastrol treatment. As shown in Figure 2E, PS-341 dramatically blocked Celastrol-triggered β-catenin degradation, but not MDL-28170. In addition, Ub-K48R, which bears a substitution of lysine 48 with arginine and blocks ubiquitin-dependent proteasomal degradation, attenuated β-catenin degradation in HEK293 cells (have wildtype Wnt components) upon Celastrol treatment, compared with wildtype ubiquitin (Figure 2F). As previous studies indicated that β-catenin degradation is initiated by phosphorylation and subsequently by the ubiquitin–proteasome system, 3 we immunoprecipitated β-catenin from SW480 and HCT116 cell lysates that were treated with or without Celastrol, β-catenin ubiquitination and phosphorylation were both significantly increased upon Celastrol treatment as shown in Figure 2G. The interaction with β-transducin repeat-containing protein (β-TrCP), a component of a dedicated E3 ubiquitin ligase complex of β-catenin, 3 was also increased by Celastrol. Whereas Celastrol decreased the interaction between β-catenin and glycogen synthase kinase-3β (GSK-3β) phosphorylation at serine 9 (Figure 2G), which inhibits GSK-3β activity to phosphorylate β-catenin and present the protein for degradation. 39 The above results indicated that Celastrol induced β-catenin degradation through the ubiquitin–proteasome system.
YAP was involved in Celastrol-induced β-catenin degradation
The Hippo pathway genetically and functionally interacts with Wnt/β-catenin pathway, especially the interaction between YAP and β-catenin.5,40 Therefore, we exogenously overexpressed wildtype YAP and mutant YAPS127A in SW480 and HCT116 cells to examine the interaction in CRC cells with Celastrol treatment. As shown in Figure 3A, Celastrol-induced β-catenin decrease could be impaired by overexpression of nuclear-localized YAPS127A, but not wildtype YAP. Surprisingly, Celastrol enhanced YAP phosphorylation at serine 127, which caused subsequent cytoplasmic translocation of YAP and induced YAP degradation (Figure 3A). As shown in Supplementary Figure S3C, the nuclear β-catenin was hardly expressed and overexpression of YAP promoted Celastrol-induced β-catenin degradation in the cytoplasm, which suggested that YAP promoted cytoplasmic but not nuclear β-catenin degradation. Attenuating YAP by siRNA in SW480 and HCT116 cells led to a remarkable block of β-catenin degradation (Figure 3B), suggesting the indispensability of YAP in β-catenin degradation.
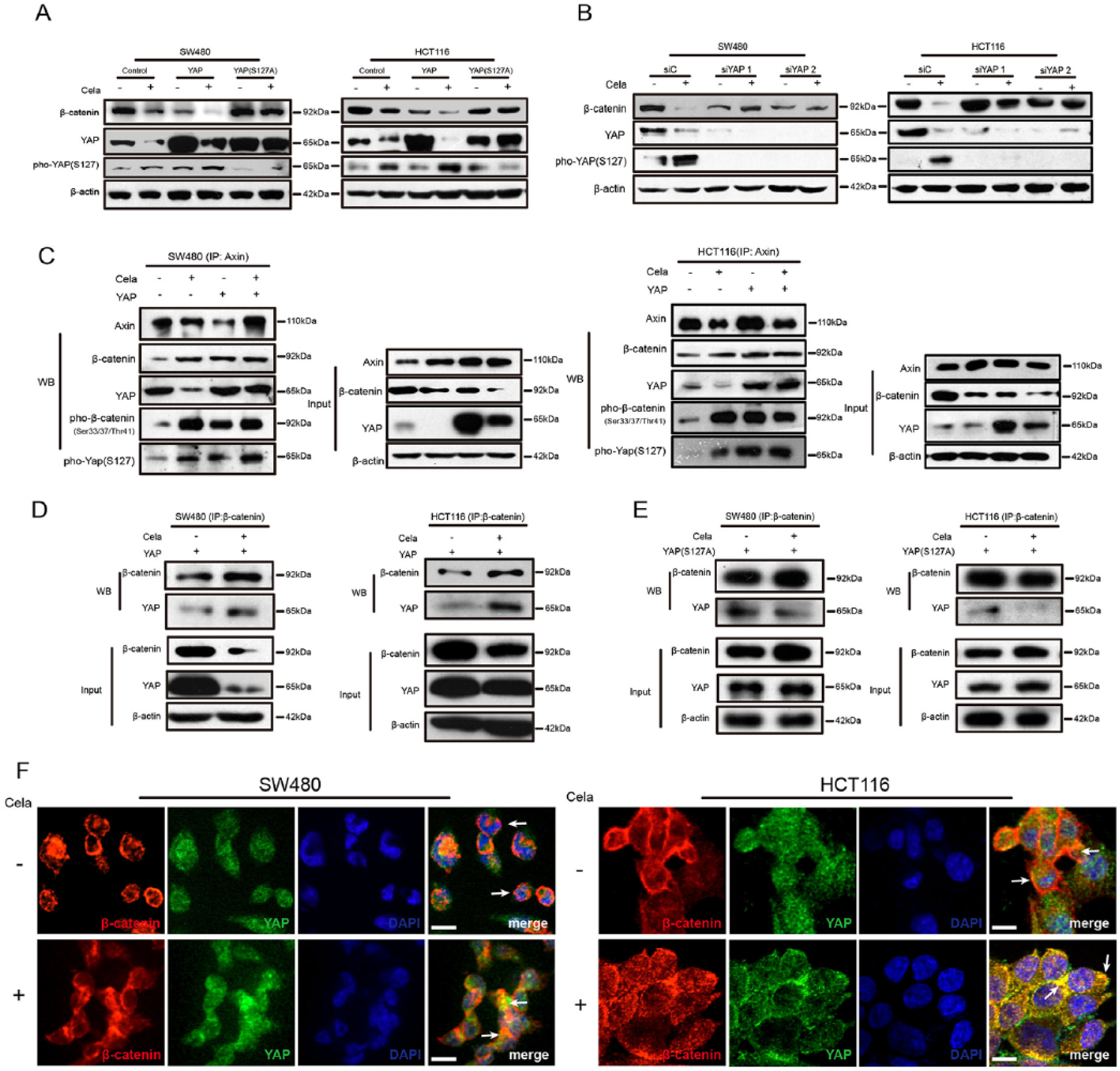
Yes-associated protein (YAP) was involved in Celastrol-induced β-catenin degradation.
To further study the association of YAP and Axin, we transfected YAP to SW480 and HCT116 cells separately, treated with Celastrol, and carried out immunoprecipitation (IP) of endogenous Axin and β-catenin, respectively, as compared with nontransfected and nontreated control cells. As shown in Figure 3C, YAP was associated with Axin and β-catenin. Exogenous YAP increased β-catenin phosphorylation. Moreover, Celastrol treatment further enhanced the association of YAP and β-catenin and YAP-induced β-catenin phosphorylation (Figure 3D). In contrast, overexpression of a mutant form of YAP (YAPS127A) reduced this association (Figure 3E). Next, we performed immunofluorescent staining to confirm the co-localization of β-catenin and YAP. β-catenin staining was detected predominantly in the nucleus of SW480 cells and in the membrane of HCT116 cells. When treated with Celastrol, the co-localization of β-catenin and YAP became much stronger by sequestering in the cytoplasm in both SW480 and HCT116 cells (Figure 3F). In addition, YAP overexpression promoted Celastrol-induced apoptosis, and mutant YAPS127A overexpression or YAP knockdown restored the effect (Supplementary Figure S3D). Taken together, our data suggested that YAP and its phosphorylation were involved in Celastrol-induced β-catenin degradation.
Celastrol-induced β-catenin degradation and YAP phosphorylation were partly mediated by activating the LKB1–AMPKα pathway via HSF1
Previous data showed that YAP phosphorylation was important in the Celastrol-induced β-catenin degradation. LKB1 represses YAP activity via promoting YAP phosphorylation, nuclear exclusion, and proteasomal degradation. 15 We found that LKB1 expression was affected by Celastrol both in vitro and in vivo (Figure 4A and B), via slowing down the degradation process (Supplementary Figure S4A and B). We exogenously overexpressed LKB1 in HCT116 and SW480 cells, and observed that LKB1 upregulation enhanced β-catenin degradation and YAP phosphorylation that was related to YAP degradation (Figure 4C). We also used the CRISPR-Cas9 system to stably establish the LKB1-knockout SW480 and HCT116 cells. The resistant monoclonal cells were designated as SW480/LKB1-KO and HCT116/LKB1-KO, respectively. Results showed that LKB1 deficiency attenuated Celastrol-induced β-catenin degradation (Figure 4D), whereas recovery of LKB1 expression in SW480/LKB1-KO and HCT116/LKB1-KO cells significantly decreased β-catenin expression (Figure 4E). These data implied that LKB1 was indispensable in promoting β-catenin degradation in CRC cells.

Celastrol-induced β-catenin degradation was dependent on LKB1.
In addition to upregulating LKB1 protein expression, Celastrol also affected LKB1 transcriptional levels. LKB1 promoter contains a predicted HSF1 binding site (Figure 5A), implying that Celastrol may enhance LKB1 transcriptional activity via HSF1. We overexpressed HSF1 in SW480 and HCT116 cells, and detected increased LKB1 mRNAs (Figure 5B). ChIP assay identified HSF1 binding to the heat shock element (HSE) on LKB1 promoter (Figure 5C). Celastrol treatment induced HSF1 expression in SW480 and HCT116 cells (Figure 5E), and elevated HSF1 also increased both LKB1 protein expression and YAP phosphorylation (Figure 5D and E). In addition, siRNA-mediated HSF1 suppression resisted β-catenin degradation through downregulation of LKB1 and YAP phosphorylation (Figure 5F).

Celastrol increased the transcriptional activation of LKB1 partly through HSF1.
The energy sensor AMPK family proteins are the main downstream targets of LKB1, 41 and AMPK also modulated YAP. 42 Celastrol-induced HSF1 upregulation also increased AMPKα phosphorylation, whereas siRNA-mediated HSF1 attenuation downregulated AMPKα phosphorylation (Figure 5E and F). Furthermore, as shown in Figure 6A, LKB1 overexpression increased AMPKα phosphorylation, but did not change the total AMPKα protein levels. Celastrol treatment further induced AMPKα phosphorylation in both SW480 and HCT116 cells. Compound C (also known as Dorsomorphin 2HCl) is an effective and reversible AMPK selective inhibitor. 43 When Compound C was used to inhibit AMPKα expression, Celastrol-induced β-catenin and YAP degradation in SW480 and HCT116 cells were blocked as compared with the nontreated group, and LKB1 overexpression could partly counteract the effect (Figure 6B), which suggested that AMPKα partially promoted Celastrol-induced β-catenin and YAP degradation in LKB1-overexpressing SW480 and HCT116 cells. Moreover, IP was performed in SW480 and HCT116 cells treated with or without Celastrol. As shown in Figure 6C, Celastrol treatment enhanced the AMPKα and LKB1 complex formation and also increased YAP phosphorylation.
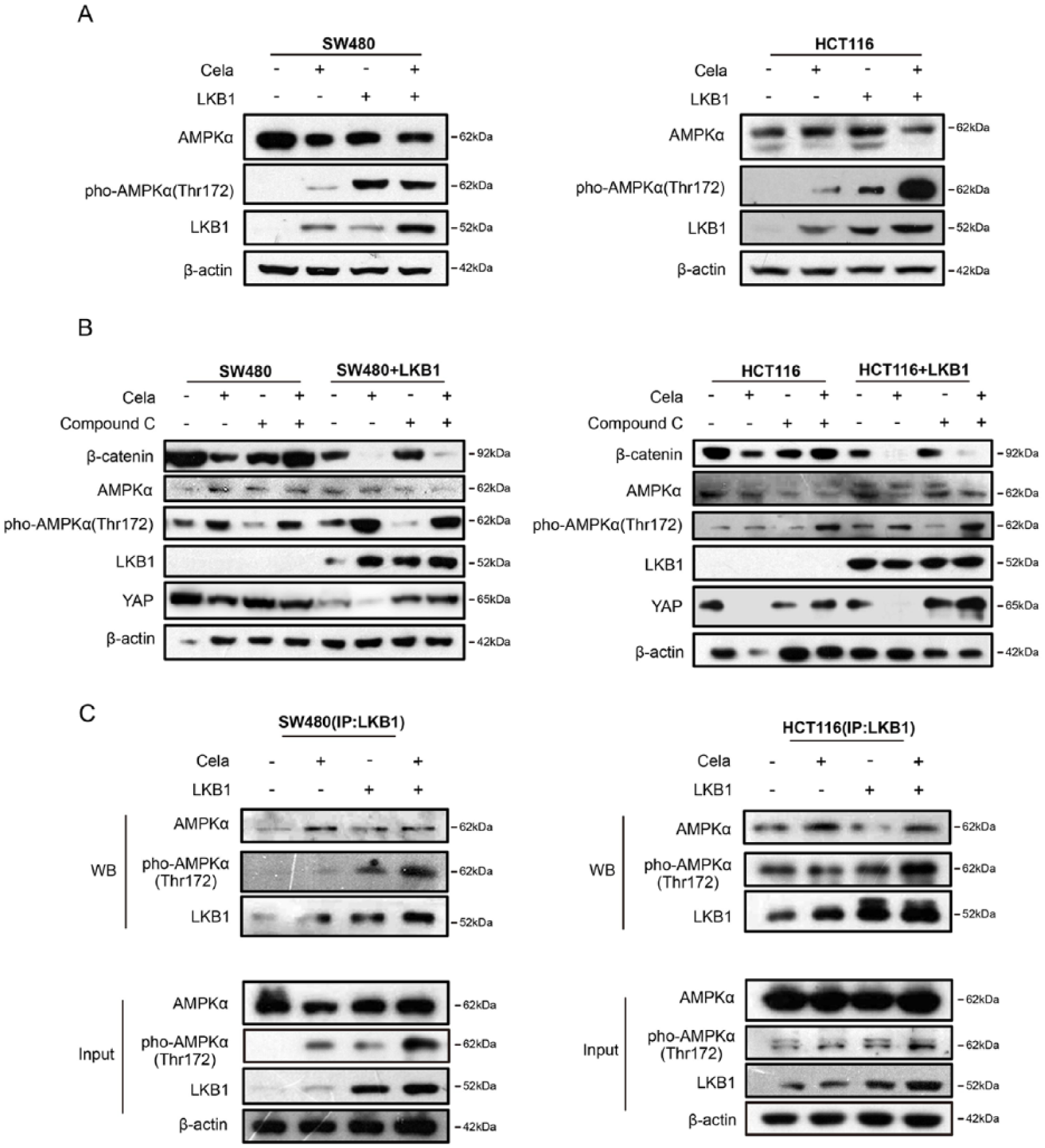
Celastrol-induced β-catenin degradation was partly mediated by activating the LKB1–AMPKα pathway.
These above findings further elucidated the molecular mechanisms of Celastrol-induced β-catenin degradation, which at least partially via the HSF1-LKB1/AMPKα–YAP cascade.
LKB1 deficiency promoted CRC progression and attenuated the tumor inhibitory effect of Celastrol
To further investigate LKB1 roles in Celastrol-mediated inhibition of colorectal tumor growth, colony formation in vitro and xenograft growth assay in vivo were performed. As shown in Figure 7A and B, LKB1 depletion in SW480/LKB1-KO and HCT116/LKB1-KO cells resulted in increased colonies in vitro and increased tumor size in vivo as compared to the parental control cells. Celastrol-mediated inhibition of colorectal cell growth in vitro and in vivo were attenuated by LKB1 depletion in SW480/LKB1-KO and HCT116/LKB1-KO cells compared with control groups, respectively. β-catenin downregulation and LKB1 upregulation were both detected in xenografts of SW480 and HCT116 cells with Celastrol treatment, as compared with nontreated control groups. However, β-catenin expression did not change in tumors derived from SW480/LKB1-KO and HCT116/LKB1-KO cells treated with or without Celastrol, and LKB1 expression was not detectable (Figure 7C).

LKB1 deficiency promoted colorectal cancer progression and attenuated the tumor inhibitory effect of Celastrol.
To better understand the roles of LKB1 in vivo, intestinal-specific LKB1-KO (Cre+ LKB1+/loxp) mice were treated with AOM/DSS and received intraperitoneal injection of Celastrol (1mg/kg) or PBS controls for 3 weeks (three times/week). Compared with the control Cre- LKB1+/loxp mice, Cre+ LKB1+/loxp mice formed more tumors with the treatment of AOM/DSS. Interestingly, Celastrol-mediated tumor suppression was not effective in the Cre+ LKB1+/loxp mice (Figure 7D). Immunohistochemistry results showed that β-catenin expression was upregulated in the colon of Cre+ LKB1+/loxp mice, no matter whether treated or not treated with Celastrol. However, Celastrol treatment decreased β-catenin expression in the Cre-LKB+/loxp mice (Figure 7E). These results verified the important role of LKB1 in Celastrol-mediated anti-CRC effect.
Discussion
Celastrol as an immunomodulator to suppress tumor necrosis factor (TNF)-α and interleukin (IL)-6 has been applied in clinical practice. 44 In recent years, the anticancer effects of Celastrol have been investigated in different cancer cell lines and animal models. Previous studies have indicated that Celastrol exerted synergistic effects with PHA-665752 and inhibited cell growth, migration and apoptosis of c-Met-deficient hepatocellular carcinoma both in vitro and in vivo, 45 which provided the therapeutic potential of Celastrol in cancer treatment. Moreira et al. indicated that Celastrol exhibited significant chemopreventive and chemosensitizing activities on doxorubicin-resistant colon cancer cells, 46 further hinting the role of Celastrol as a candidate for adjuvant medicine on the colon cancer. In the present study, our results showed that the intraperitoneal injection of Celastrol had significantly potent inhibition effect on spontaneous and carcinogen-induced colitis-associated CRC in mice, and the inhibitory efficiency of Celastrol is similar to 5-FU (Supplementary Figure S1B and C). Interestingly, another study indicated that dietary Celastrol supplementation also had the suppressive effect on CRC, 47 which further suggested the potential for Celastrol as a clinical medication. To further understand the mechanism of Celastrol in inhibiting CRC growth would be beneficial for the therapeutic application of Celastrol. Bufu et al. indicated that Celastrol-induced anti-tumor effects were mediated through MMP3 and MMP7 by the PI3K/AKT signaling pathway, which is crucial for CRC development and progression. 48 However, dysregulated APC/β-catenin signaling pathway is an early and generally accepted event in CRC tumorigenesis. Here, we demonstrated that Celastrol promoted β-catenin degradation in vitro and in vivo. Sustained activation of β-catenin facilitated colony formation of SW480 and HCT116 cells. Celastrol decreased β-catenin abundance in a dose- and time-dependent manner in SW480 and HCT116 cells (Figure 2A and Figure S2A), and also suppressed downstream genes of β-catenin, including c-Myc, survivin, CYR61, and Cyclin D1.
Downregulation of Hippo signaling is correlated with upregulation of Wnt/β-catenin signaling in human CRCs. The tumor-derived stabilized form of β-catenin induced by the mutant APC was suppressed by activation of Hippo pathway. 40 YAP/TAZ act as downstream effectors of the alternative Wnt signaling pathway and mediate the biological functions including gene expression, osteogenic differentiation, cell migration, and antagonism of Wnt/β-catenin signaling. 49 However, some studies indicated that YAP was essential for β-catenin nuclear accumulation in intestinal regeneration and survival of β-catenin-driven CRC.9,10 Here, we found that phosphorylated YAP was required in the process of Celastrol-induced β-catenin degradation. Phosphorylated YAP loses its activation by sequestering in the cytoplasm and is degraded through the ubiquitin–proteasome pathway. 50 YAPS127A causes YAP accumulation from the cytoplasm into the nucleus without degradation. Our data showed that overexpressing the wildtype YAP intensified Celastrol-induced β-catenin degradation, whereas the nuclear-localized YAPS127A prevented β-catenin degradation (Figure 3A). Recent studies pointed out that Ser397 of YAP could regulate the location of YAP in the cytoplasm and nucleus51,52 and regulate YAP degradation. Our results also indicated that Celastrol and LKB1 overexpression could induced the phosphorylation at S397 site of YAP (data not shown). These may provide hints that the phosphorylation at S127 could cause YAP sequestering in the cytoplasm, which promoted the degradation of β-catenin through the destruction complex, however, the degradation of YAP may need other phosphorylated sites, such as S397.
In addition, our data proved that LKB1 played an important role in Celastrol-mediated inhibition of CRC growth. LKB1 is commonly known as a tumor suppressor gene because its hereditary mutation is responsible for Peutz–Jeghers syndrome, 53 and somatic inactivation of LKB1 is found in non-small cell lung cancer, melanoma, and cervical cancers. 54 Therapeutic strategies against LKB1-mutant cancer are emerging. 54 We showed that LKB1 deficiency promoted CRC cell growth and partially attenuated the inhibitory effect of Celastrol in tumor growth both in vitro and in vivo. Notably, the homozygous deletion of LKB1 (Cre+ LKB1loxp/loxp) mice form more tumors in colon with the treatment of AOM/DSS than the Cre+ LKB1+/loxp mice, and Celastrol almost has no effect on the colon tumor growth in Cre+ LKB1loxp/loxp mice (data not shown). AMPKα, the downstream effector of LKB1, also participated in the process of Celastrol-induced β-catenin degradation. Celastrol influenced the phosphorylation of AMPK, but not MAPK (data not shown). The inhibitor of AMPK partially inhibited Celastrol-induced β-catenin and YAP degradation, which further suggested the connection between LKB1, AMPK, and YAP.
Furthermore, intestinal inflammation is inseparable with colitis-associated cancer (CAC). 55 As a traditional herbal drug, Celastrol has been used to treat inflammatory disease such as allergic asthma. 19 The latest research showed that Celastrol results in appetite reduction (but not reduction in energy expenditure) and dramatic weight loss in hyperleptinemic diet-induced obese (DIO) mice by increasing leptin sensitivity, but not in mice that lack leptin actin, 56 which demonstrated that Celastrol is a true leptin sensitizer.56,57 More interestingly, Lei Cao et al. 58 found that enriched environment (EE) could significantly reduce cancer burden in both a syngeneic melanoma as well as a colon cancer model through hypothalamic brain-derived neurotrophic factor (BDNF)/leptin axis via sympathoneural β-adrenergic signaling. All these experimental findings pointed out the relationship between the central nervous system (CNS) and cancer and suggested that Celastrol as a promising agent for the pharmacological treatment of colon cancer may have other mechanisms. More studies are needed to identify the mechanism of action of Celastrol.
There are some limitations in our study. First, our study showed that Celastrol-induced β-catenin degradation was dependent on the phosphorylation of YAP S127. Whether YAP phosphorylation at S127 site involved in Celastrol treatment and LKB1/AMPK signaling was essential for YAP degradation is worth to further investigated in future studies. Second, we fleshed out the molecule-to-molecule mechanism of Celastrol-induced β-catenin degradation in animal and cellular models. To expand the application of Celastrol as a preclinical cancer drug, we will need to further verify the effect of Celastrol in patient-derived xenograft (PDX) models of CRC.
In summary, our current study first revealed that Celastrol inhibited CRC cell growth in vitro and in vivo by promoting β-catenin degradation, through activating the LKB1–AMPKα pathway and phosphorylating YAP. Celastrol upregulated HSF1 expression, and HSF1 overexpression enhanced LKB1 transcriptional activity. Subsequently, LKB1 activated AMPKα and YAP, and promoted β-catenin degradation through the ubiquitin–proteasome system (Figure 7F). These observations elucidate the molecular mechanisms of the effect of Celastrol on CRC growth, and provide evidence in support of the potential application of Celastrol in CRC therapy.
Supplemental Material
Figure_S1_(1) – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S1_(1) for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S2_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S2_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S3_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S3_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S4_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S4_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S5_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S5_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S6_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S6_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Figure_S7_for_modification – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Figure_S7_for_modification for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Supplementary_material-revision_(1) – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Supplementary_material-revision_(1) for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Supplemental Material
Table_S1_(1) – Supplemental material for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer
Supplemental material, Table_S1_(1) for LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer by Shuren Wang, Kai Ma, Cuiqi Zhou, Yu Wang, Guanghui Hu, Lechuang Chen, Zhuo Li, Chenfei Hu, Qing Xu, Hongxia Zhu, Mei Liu and Ningzhi Xu in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors thank Professor Quan Chen for the parental T-Rex-293 cells.
Author contributions
Conception and design: Shuren Wang, Mei Liu, Ningzhi Xu.
Acquisition of data: Shuren Wang, Kai Ma, Yu Wang, Guanghui Hu.
Analysis and interpretation of data: Shuren Wang, Mei Liu.
Administrative, technical, or material support: Lechuang Chen, Zhuo Li, Cuiqi Zhou, Chenfei Hu, Qing Xu, Hongxia Zhu.
Project administration: Shuren Wang, Mei Liu, Ningzhi Xu.
Study supervision: Mei Liu, Ningzhi Xu.
Writing original draft: Shuren Wang.
Writing review and editing: Shuren Wang, Mei Liu, Ningzhi Xu, Cuiqi Zhou.
Funding
This work was supported by the National Key Research and Development (R&D) Program of China (grant numbers 2016YFC0906000, 2016YFC0906002, and 2016YFC1302103), National Natural Science Foundation (grant number 81321091), CAMS Innovation Fund for Medical Sciences (CIFMS) (grant number 2016-I2M-1-001), National Basic Research Program of China (grant number 2011CB910704), and Postgraduate Innovation Fund of the Beijing Union Medical College (grant number 2014-0710-1015), P. R. China. The funders had no role in study design, data collection and analysis, interpretation of date, or preparation of the manuscript.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.