
Abstract
Background:
Capecitabine is generally dosed based on body surface area (BSA). This dosing strategy has several limitations; however, evidence for alternative strategies is lacking. Therefore, we analyzed the toxicity and effectiveness of fixed-dose capecitabine and compared this strategy with a BSA-based dose of capecitabine in a large set of patients.
Methods:
Patients treated with fixed-dose capecitabine between 2003 and 2015 were studied. A comparable group of patients, dosed based on BSA, was chosen as a control cohort. A total of two combined scores were used: capecitabine-specific toxicity (diarrhea, National Cancer Institute Common Toxicity Criteria grade ⩾3, hand-foot syndrome ⩾2, or neutropenia ⩾2), and clinically relevant events due to toxicity, that is, hospital admission, dose reduction, or discontinuation. Per treatment regimen, patients were divided into three BSA groups based on BSA quartiles corrected for sex. Toxicity scores were compared by a Chi-square test between cohorts, and within cohorts using BSA groups. Progression-free survival (PFS) was estimated by the Kaplan–Meier method.
Results:
A total of 2319 patients were included (fixed dosed, n = 1126 and BSA-based dose, n = 1193). Overall, four regimens were evaluated: capecitabine-radiotherapy (n = 1178), capecitabine-oxaliplatin (n = 519), capecitabine triplet (n = 181) and capecitabine monotherapy (n = 441). The incidence of capecitabine-specific toxicity and clinically relevant events was comparable between fixed-dose and BSA-dosed patients, while a small difference (7.1%) in absolute dose was found. Both cohorts showed only a higher incidence of both toxicity scores in the lowest BSA group of the capecitabine-radiotherapy group (p < 0.05). Subgroups of the fixed-dose cohort analyzed for PFS, showed no differences between BSA groups.
Conclusions:
Fixed-dose capecitabine is as comparably well tolerated and effective as BSA-based dosing and could be considered as a reasonable alternative for BSA-based dosing.
Introduction
Capecitabine is an oral prodrug for the cytotoxic agent 5-fluorouracil (5-FU) and is widely used in the treatment of colorectal cancer and other solid tumors (e.g. breast and gastric cancer).1–4 Capecitabine has equal effectiveness and shows, in general, a more favorable toxicity profile compared with intravenous 5-FU, except for the incidence of hand-foot syndrome (HFS). 5 Depending on the different types of treatment regimens, capecitabine is given either as monotherapy, in combination with other cytotoxic agents, or it is combined with radiotherapy. Worldwide, dosing of capecitabine for the individual patient is based on the patient’s body surface area (BSA). However, both effectiveness and toxicity depend on the individual exposure to capecitabine and therefore the rationale for dosing based solely on height and weight has been questioned for decades.6–13
BSA-guided dosing of anticancer agents aims to minimize inter-individual variability in exposure as a result of differences in body composition, thereby trying to achieve more similar exposure across patients, resulting in a maximal effect and limited toxicity. 14 However, this dosing strategy has several drawbacks. Firstly, there is limited evidence for the basis of the BSA formula, since the first formula to calculate BSA (by Du Bois and Du Bois, more than a century ago) was based on only nine individuals. 15 Still, it forms the backbone for all (other) BSA formulae, of which the Mosteller derivative is currently the most frequently used. 16 Secondly, BSA-based dosing has increased costs and a larger chance of calculation errors compared with fixed dosing. 6 Thirdly and most importantly, although BSA dosing was intended to reduce the inter-individual variability in drug exposure, many researchers have concluded that for the majority of anticancer agents there is no clear relationship between an individual’s exposure and a BSA-based dose.7–13 Indeed, Baker and colleagues demonstrated by modeling that inter-individual variability in the clearance of capecitabine expressed as coefficient of variation was increased when BSA was taken into account (31.3% versus 36.5%). 10 In other words, there is fair skepticism regarding the question of whether this dosing strategy really contributes to reducing inter-individual pharmacokinetic and consequent pharmacodynamic variability of anticancer agents.11,13
BSA-based dosing is for many anticancer agents not evidence based, and especially for frequently used drugs such as capecitabine, there is a need for alternative dosing strategies to standardize the dose. 17 Fixed dosing means that the dose is not adjusted for body size, so that every adult patient with the same malignancy receives the same (fixed) dose. A major benefit of dose standardization by fixed dosing is that it will lead to fewer prescribing errors and a reduction in preparation and storage costs.18–20 Fixed dosing is already implemented in the majority of newly developed oral anticancer drugs. 21 However, unless there is more evidence that a fixed dose can safely be applied without compromising effectiveness, then conventional chemotherapy regimens will continue to be dosed based on BSA according to the registration studies, even though BSA-guided dosing is in many cases not evidence based.
To the best of our knowledge, there are no published studies with a sufficiently large sample size evaluating the outcomes of a fixed dose of capecitabine. In 2003, the Erasmus University Medical Center, Rotterdam, the Netherlands, implemented a fixed dose of capecitabine in different treatment regimens, as there was no evidence that BSA-based dosing was better. This resulted in a unique ‘real-life’ cohort of patients treated with a fixed dose of capecitabine, with a long follow-up period. Therefore, the aim of our present study was to evaluate the toxicity and effectiveness of fixed-dose capecitabine in several treatment schedules in this cohort of patients. Additionally, we compared this cohort with another large cohort of Dutch cancer patients, in which patients were dosed based on BSA and treated in the same time period, in order to determine whether fixed-dose capecitabine is as equally well tolerated and effective as BSA-based dosing of capecitabine.
Methods
The cohorts for this analysis were obtained from the Erasmus University Medical Center, Rotterdam, the Netherlands, for the fixed-dose cohort, and from the Netherlands Cancer Institute, Antoni van Leeuwenhoek, Amsterdam; Slotervaart Hospital, Amsterdam; and Canisius Wilhelmina Hospital, Nijmegen, all in the Netherlands for the BSA-based dose cohort, respectively.
The primary study endpoint was the incidence of treatment-related toxicity in a fixed-dose cohort compared with a BSA-based dose cohort. Secondary endpoints included the comparison of the absolute amount of capecitabine administered in the fixed-dose cohort compared with BSA-dosing strategies, incidence of toxicity between BSA groups within both cohorts, and the effectiveness of fixed-dose capecitabine compared between BSA groups in terms of disease-free survival (DFS) in (neo)adjuvant care and progression-free survival (PFS) in palliative care.
Patients and treatments
All patients treated with capecitabine between 2003 and 2015 at the Erasmus University Medical Center were identified by the hospital pharmacy based on drug dispensing data and evaluated for inclusion in the fixed-dose cohort. As fixed-dose capecitabine is considered routine clinical care at the Erasmus University Medical Center, no ethics approval or informed consent was required to retrospectively collect and analyze these patient data for research purposes. Patients in the BSA-based dose cohort were prospectively included in a previously conducted trial in three large hospitals in the Netherlands (ClinicalTrials.gov identifier: NCT00838370), this trial was approved by the medical ethical committees of all participating hospitals. 22 All patients of the BSA-based dose cohort provided informed consent for the prospective trial, including consent for additional analyses outside the subject of this trial. Patients were excluded from both cohorts when they had a World Health Organization (WHO) performance status of 3 or 4, when they were previously treated with fluoropyrimidines, when they were treated in an experimental treatment setting outside the standard-of care; or when limited data on important parameters required for the current analysis were available (i.e. length, weight, toxicity evaluation).
In both cohorts, patients were divided into four groups based on the treatment regimen: capecitabine monotherapy (CAPE MONO), capecitabine combined with radiotherapy (CAPE + RT), capecitabine combined with oxaliplatin (CAPOX), and capecitabine triplet therapy (CAPE TRIPLET). The CAPE TRIPLET group consisted of capecitabine combined with epirubicin and cisplatin or oxaliplatin (ECC/EOX) in both cohorts, and in the BSA-based dose cohort also patients treated with capecitabine with docetaxel and oxaliplatin (DOC) were included. Capecitabine was administered as a fixed daily dose (divided over two doses daily) of 3000 mg for CAPE + RT; 3500 mg for CAPOX and ECC/EOX; 3500 mg or 4000 mg for CAPE MONO. Detailed descriptions of included treatment types are given in Supplementary Table 1.
Data
All data for the fixed-dose cohort were retrospectively collected from the electronic health records. For the BSA-based dose cohort all data was prospectively collected in a previously conducted trial by Deenen and colleagues. 22 Data on patient demographics (i.e. length, weight, WHO performance status) had to be known within 1 month before the start of capecitabine. BSA was calculated per patient using the Mosteller formula. 16 Renal function was expressed as estimated glomerular filtration rate (eGFR). For the fixed-dose cohort the eGFR was calculated according to the Cockcroft–Gault formula, 23 and for the BSA-based dose cohort according to the Modification of Diet in Renal Disease formula. 24 Toxicity was defined as all possible capecitabine-related adverse events and laboratory abnormalities occurring during treatment with capecitabine until 1 month after the end of treatment or until the start of a new treatment, whichever occurred first. Toxicity was graded according to the Common Terminology Criteria for Adverse Events (CTC-AE) version 4.03. 25 Overall, two combined scores were created to evaluate severe toxicity and clinically relevant events due to toxicity. Capecitabine-specific toxicity was defined as toxicity grade for diarrhea ⩾ 3, HFS ⩾ 2, or neutropenia ⩾ 2. Clinically relevant events consisted of hospital admission, dose reduction, or discontinuation caused by possible capecitabine-related adverse events. Data on DFS in (neo)adjuvant treated patients or PFS in palliative treated patients were collected to assess the effectiveness of fixed-dose capecitabine. DFS was defined as the time till disease recurrence. PFS was defined as time till disease progression or death from any cause. Disease recurrence or progression had to be pathologically proven or by imaging, evaluated according to the response evaluation criteria in solid tumors (RECIST) 1.1. 26
Statistical analysis
Demographic characteristics were compared between the two cohorts by using the Chi-square test for categorical variables, and an unpaired Student’s t test or Mann–Whitney U test for continuous variables. Per treatment, toxicity was compared between the fixed-dose and BSA-based dose cohort using the Chi-square or Fisher’s exact test. In the fixed-dose cohort, patients were divided into three groups based on BSA quartiles per sex and treatment: lowest 25%, middle 50% and highest 25%. In the BSA-based dose cohort, patients were divided into the same treatment groups based on the BSA limits per sex obtained from the fixed-dose cohort. Toxicity was compared between the three BSA groups within both cohorts using the Chi-square test for trend.
For regimens where BSA was found to be predictive for toxicity, other relationships between known risk factors from the literature and toxicity were studied within both cohorts using univariate and multivariate binary logistic regression analysis, where the assumption of linearity was checked for each continuous risk factor. Significant risk factors with p < 0.05 detected in the univariate analysis of the fixed-dose cohort, were included in the multivariate analysis of both cohorts. In the fixed-dose cohort, the mean given fixed daily capecitabine dose was compared with a calculated (‘fictional’) mean daily capecitabine dose based on the patient’s BSA and according to clinical guidelines per treatment type, by using a paired sample Student’s t test.
Survival analysis was only performed in the fixed-dose cohort in separate groups per tumor type, indication and treatment regimen [i.e. CAPE + RT for locally advanced colorectal cancer (laCRC), CAPOX for metastatic colorectal cancer (mCRC), and ECC/EOX for gastric cancer]. For the BSA-based dose cohort, this analysis could not be performed because these data were not collected. Survival analysis between three BSA groups was done by the Kaplan–Meier method. Only treatment regimens per indication with at least 20 events were included in this analysis. p-values < 0.05 were considered statistically significant. All statistical analyses were performed using SPSS (version 24.0.0.1).
Results
Patient and treatment characteristics
A total of 3583 patients were screened for inclusion, of whom 1264 patients were excluded, mainly because of previous fluoropyrimidine treatment (Figure 1). This resulted in a total of 2319 patients enrolled in the analysis, of whom 1126 patients were included in the fixed-dose cohort and 1193 patients in the BSA-based dose cohort (Figure 1). Patient characteristics for both cohorts per treatment group are described in Table 1. Overall, more male patients were included in the fixed-dose cohort (61%) than in the BSA-based dose cohort (48%; p < 0.001). The mean age was comparable in most treatment groups, but patients treated with capecitabine monotherapy were slightly older in the fixed-dose cohort (65 versus 61 years, p = 0.019). The majority of patients were of White origin (91%), but fewer were in the fixed-dose cohort (85%) compared with the BSA-based dose cohort (96%; p < 0.001). The BSA of patients was normally distributed per sex and treatment. The mean BSA of patients was comparable in most treatment groups, but in the CAPE + RT group the BSA was slightly higher in the fixed-dose cohort compared with the BSA-based dose cohort (1.94 m2 and 1.91 m2, respectively, p = 0.013).

STROBE diagram of included patients.
Patient characteristics.

BSA was calculated according to the Mosteller formula. 16
eGFR was calculated according to the Cockcroft–Gault formula in the fixed-dose cohort, 23 and calculated according to the CKD-EPI formula in the BSA-based dose cohort. 24
The administered treatment regimens are described in more detail in Supplemental Table 1.
Total daily capecitabine dose at start of first cycle.
BC, breast cancer; BSA, body surface area; CAPE, capecitabine; CAPOX, capecitabine combined with oxaliplatin; CRC, colorectal cancer; ECC, capecitabine combined with epirubicin and cisplatin; eGFR, estimated glomerular filtration rate; EOX, capecitabine combined with epirubicin and oxaliplatin; GC, gastric cancer; IQR, interquartile range; mono, monotherapy; SD, standard deviation; RT, radiotherapy.
p-values < 0.05 are considered statistically significant and are depicted in bold.
Overall, the most common tumor type was colorectal cancer (75%), and capecitabine combined with radiotherapy was the most often used treatment regimen in both cohorts. The median capecitabine daily dose was 3000 mg in the fixed-dose cohort, and 3500 mg in the BSA-based dose cohort. Only in the CAPE MONO group, was no significant difference in the median capecitabine daily dose found between both cohorts. Overall, the mean given fixed dose of capecitabine was 7.2% lower than the calculated dose based on BSA (p < 0.001); the results detailed per treatment are shown in Supplementary Table 2.
Toxicity between the fixed-dose and BSA-based dose cohort
No differences in the incidence of capecitabine-specific toxicity or clinically relevant events could be identified between the fixed-dose and BSA-based dose cohort per treatment group (Table 2). Only in the CAPE MONO and CAPE TRIPLET groups were some minor differences in the single toxicity incidences identified. In the fixed-dose patients of the CAPE MONO group, a lower incidence of HFS ⩾ 2 (22% versus 33%, p = 0.026) and a higher incidence of neutropenia ⩾ 2 (14% versus 6%, p = 0.005) was observed than in the BSA-based dose patients (Table 2). Fixed-dose patients of the CAPE TRIPLET group had a higher incidence of neutropenia ⩾ 2 (82% versus 61%, p = 0.003), and more discontinuation of treatment due to toxicity (37% versus 23%, p = 0.043) compared with the BSA-based dose patients (Table 2). Importantly, no difference in toxicity or clinically relevant events could be identified when toxicity was compared between the lowest BSA quartile of the fixed-dose and BSA-based dose cohort per treatment, indicating that patients with a low BSA did not receive too much capecitabine in the fixed-dose cohort (Supplementary Table 3).
Toxicity compared between fixed-dose and BSA-based dose patients per treatment regimen.

Capecitabine-specific toxicity was defined as at least one of the following toxicity scores: diarrhea ⩾ 3, HFS ⩾ 2, neutropenia ⩾ 2.
Clinically relevant events were defined as at least one of the following events due to toxicity: dose reduction, stop with capecitabine, hospital admission.
The administered treatment regimens are described in more detail in Supplemental Table 1.
BSA, body surface area; CAPE, capecitabine; CAPOX, capecitabine combined with oxaliplatin; HFS, hand-foot syndrome; mono, monotherapy; RT, radiotherapy.
p-values < 0.05 are considered statistically significant and are depicted in bold.
Toxicity compared between BSA groups within both cohorts
No differences could be identified between the CAPOX, CAPE MONO and CAPE TRIPLET groups when the incidence of capecitabine-specific toxicity and clinically relevant events was compared between the low, middle and high BSA group per treatment and cohort. However, only in the CAPE + RT group a significant difference in capecitabine-specific and clinically relevant events could be identified between BSA groups within the fixed-dose cohort (p = 0.009 and p = 0.013, respectively) and within the BSA-based dose cohort (p = 0.022 and p = 0.035, respectively; Table 3), demonstrating a higher risk of toxicity in the lowest BSA quartile of patients from the CAPE + RT group of both cohorts.
Toxicity compared between BSA groups within the fixed-dose cohort and the BSA-based dose cohort.
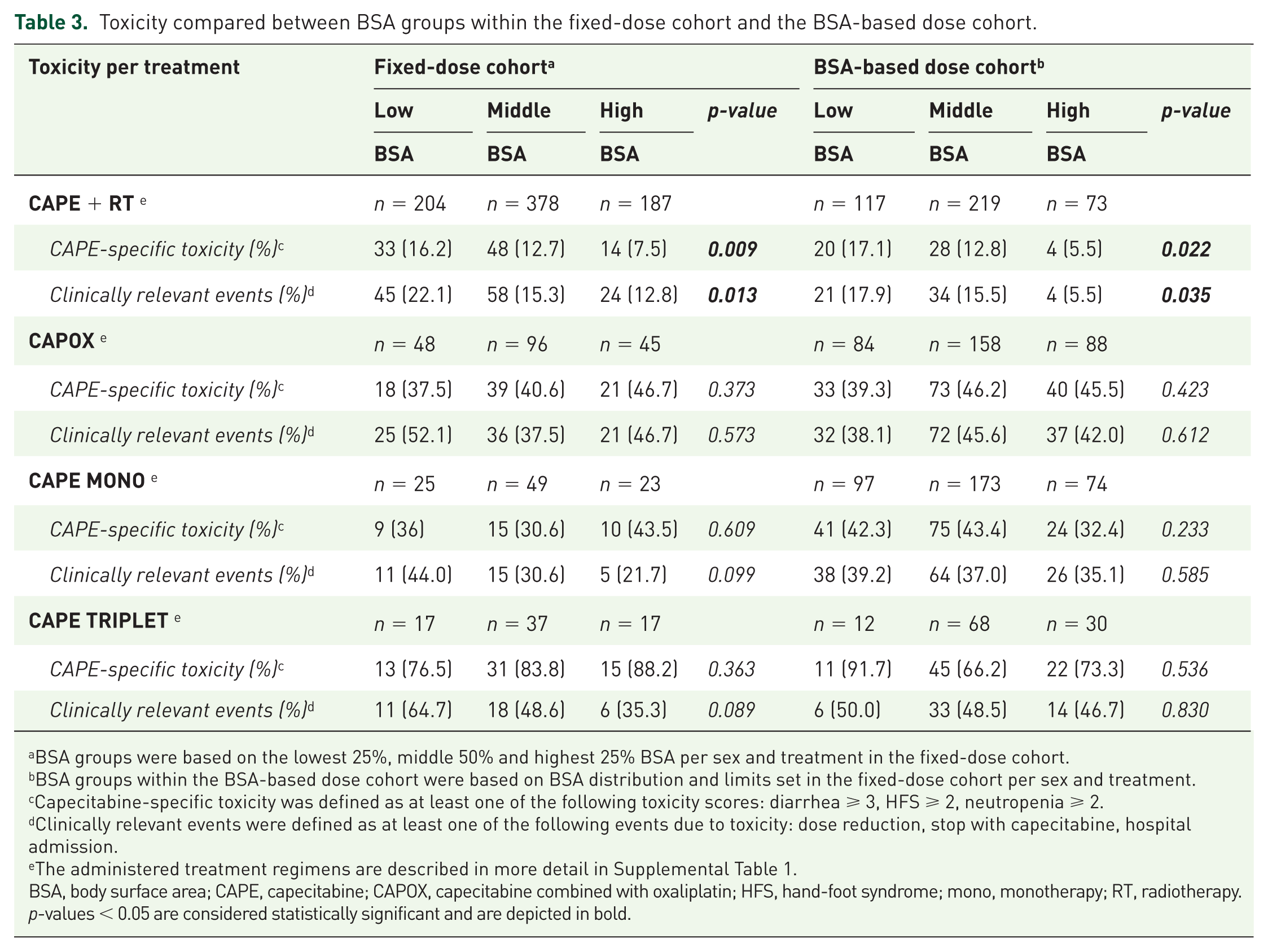
BSA groups were based on the lowest 25%, middle 50% and highest 25% BSA per sex and treatment in the fixed-dose cohort.
BSA groups within the BSA-based dose cohort were based on BSA distribution and limits set in the fixed-dose cohort per sex and treatment.
Capecitabine-specific toxicity was defined as at least one of the following toxicity scores: diarrhea ⩾ 3, HFS ⩾ 2, neutropenia ⩾ 2.
Clinically relevant events were defined as at least one of the following events due to toxicity: dose reduction, stop with capecitabine, hospital admission.
The administered treatment regimens are described in more detail in Supplemental Table 1.
BSA, body surface area; CAPE, capecitabine; CAPOX, capecitabine combined with oxaliplatin; HFS, hand-foot syndrome; mono, monotherapy; RT, radiotherapy.
p-values < 0.05 are considered statistically significant and are depicted in bold.
Risk factors for toxicity in patients treated with CAPE + RT
Only in the CAPE + RT group, an increased toxicity risk was demonstrated in the low BSA group in both cohorts. Therefore, in this group additional analyses for other risk factors than BSA were performed. Univariate regression analysis, demonstrated that BSA was predictive for toxicity in the CAPE + RT group of both cohorts (Table 4). Sex, age, and kidney function were also significantly related to toxicity in the fixed-dose cohort, but not in the BSA-based dose cohort. After correction for these factors in a multivariate model, BSA remained significantly predictive for capecitabine-specific toxicity in the fixed-dose patients [odds ratio (OR) = 0.25, 95% confidence interval (CI) = 0.07–0.86, p = 0.028] and BSA-based dose patients (OR = 0.09, 95% CI = 0.01–0.74, p = 0.025). Interestingly, fixed-dose women treated with CAPE + RT (and the same diagnosis) had a doubling of the toxicity risk compared with men for both capecitabine-specific toxicity (OR = 2.02, 95% CI = 1.22–3.37, p = 0.007) and clinically relevant events (OR = 2.12, 95% CI = 1.35–3.32, p = 0.001; Table 4).
Risk factors for toxicity in patients treated with capecitabine and radiotherapy within the fixed-dose cohort and the BSA-based dose cohort.

Capecitabine-specific toxicity was defined as at least one of the following toxicity scores: diarrhea ⩾ 3, HFS ⩾ 2, neutropenia ⩾ 2.
Clinically relevant events were defined as at least one of the following events due to toxicity: dose reduction, stop with capecitabine, hospital admission.
BSA, body surface area; CAPE, capecitabine; CI, confidence interval; eGFR, estimated glomerular filtration rate; F, female; HFS, hand-foot syndrome; OR, odds ratio; M, male; RT, radiotherapy.
p-values < 0.05 are considered statistically significant and are depicted in bold.
Effectiveness
Overall, for mCRC patients treated with CAPOX, the median PFS was 8.6 months (95% CI 6.9–10.3 months) and for patients with gastric cancer treated with ECC/EOX, the median PFS was 24.6 months (95% CI 6.1–43.0 months). For patients with laCRC treated with CAPE + RT, the median DFS was not reached; the 5-year survival probability was 0.56. No statistical differences between BSA groups in PFS for CAPOX for mCRC or ECC/EOX for gastric cancer, nor for the DFS with CAPE + RT for laCRC, could be identified (Figure 2). These results indicate that, in the fixed-dose regimens evaluated, there was no inadequate dosing of patients. Other fixed-dose treatment regimens could not be evaluated for survival due to too low number of events per treatment and indication.
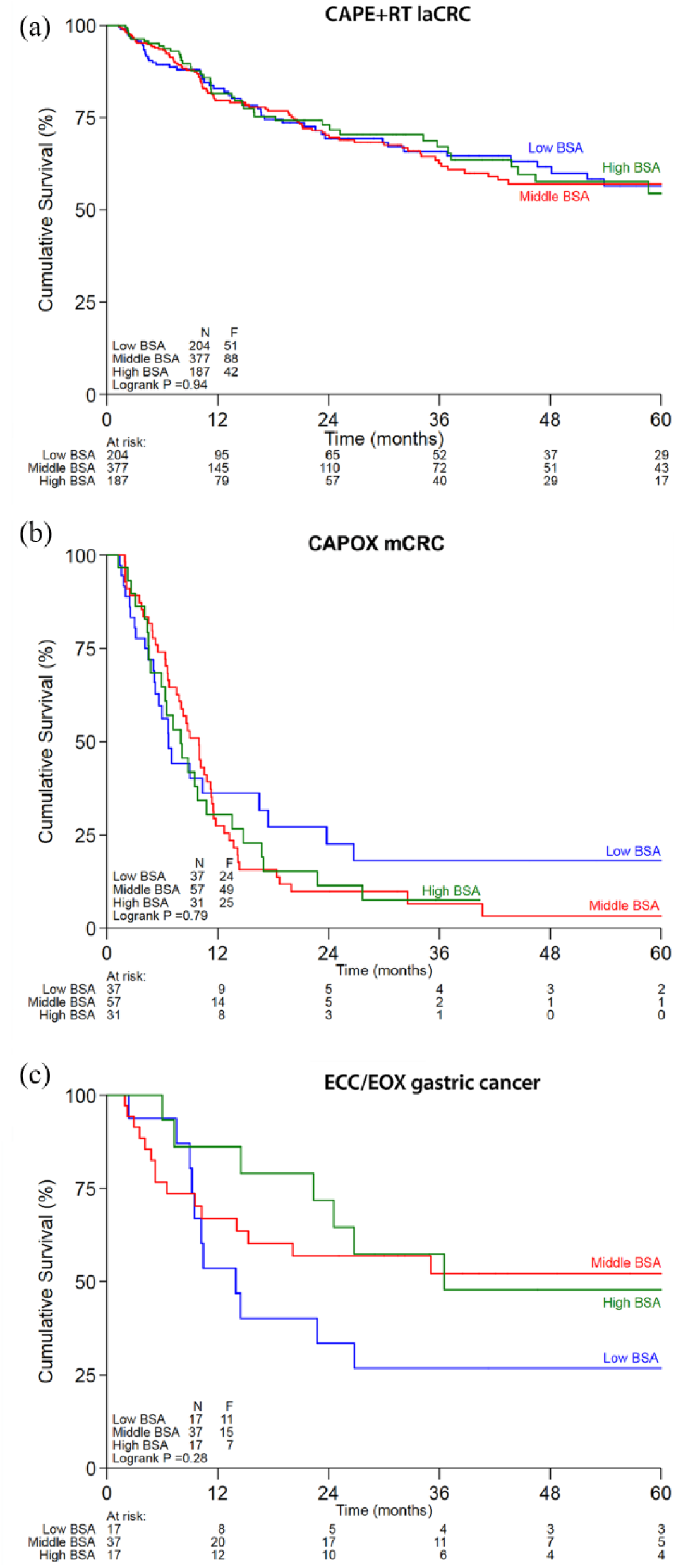
Survival compared between BSA groups within the fixed-dose cohort.
Discussion
This relatively large cohort study demonstrates that a fixed dose of capecitabine is as comparably well tolerated as dosing based on BSA in several treatment regimens (i.e. CAPOX, CAPE TRIPLET and CAPE MONO). Only in the CAPE + RT group, was a low BSA predictive for capecitabine-specific toxicity and clinically relevant events. In addition, our data suggest that a fixed dose of capecitabine was equally effective compared with dosing based on BSA. Therefore, we demonstrated that this fixed dosing strategy of capecitabine is feasible in a large ‘real-life’ population with common treatment regimens.
Beforehand, the observed association between BSA and toxicity when capecitabine was combined with radiotherapy was not expected. It is remarkable that an increased incidence of capecitabine-specific and clinically relevant events was not only found in the low BSA group in the fixed-dose cohort, but also in the low BSA group in the BSA-based dose cohort (Table 3). The fact that also in the latter group a higher risk of toxicity was observed in the lowest BSA quartile of patients, suggests that this effect is likely to be caused by the interaction of the two treatment modalities. When capecitabine is combined with radiotherapy, the absolute dose of capecitabine used is lower compared with the other regimens, because it is used as radiosensitizer. The enzyme thymidine phosphorylase in the tumor tissue is responsible for the final metabolic step in the conversion of capecitabine into 5-FU. This conversion is boosted by radiotherapy and therefore mostly local effects of 5-FU will be seen. 27 Occurrence of diarrhea during RT could be explained by (at least) two reasons. The first reason is that there is a clear relationship between radiated small bowel volume and the incidence of diarrhea in chemoradiotherapy for rectal tumors, and possibly, this is also related to BSA, since hypothetically a higher small bowel volume is exposed to radiotherapy in patients with a low BSA.28,29 Another reason is a possible relationship with rectal irritation by the tumor itself. 30 Finally, in the CAPE + RT group of both cohorts, diarrhea was the most frequent severe adverse event with an incidence of 10%. As a result, this finding might be biased because of diarrhea being the major side effect of radio-therapy. Unfortunately, no studies have been performed on the mechanism of toxicity related to capecitabine combined with radiotherapy. Further research should therefore be conducted on the potential effects of radiotherapy and BSA on toxicity of this combination treatment.
Several factors are known to influence the risk of toxicity caused by capecitabine. Older age, female sex and a decreased renal function have been related to the risk of toxicity.31–33 In our multivariate analysis in the CAPE + RT group of fixed-dose patients, we have also confirmed an increased risk of toxicity with female sex, but we could not clearly confirm the role of age and renal function. This could potentially be explained by the limited range of these two factors. In addition, in the BSA-based dose cohort all these risk factors could not be confirmed in the CAPE + RT group.
Secondly, another factor that strongly influences the risk of toxicity is genetic variation in capecitabine metabolism. The enzyme dihydropyrimidine dehydrogenase (DPD) is largely responsible for the inactivation of 5-FU, and with a decreased activity of this enzyme related to polymorphisms in the DPYD gene, the risk of severe toxicity largely increases.34,35 Only recently, genotyping of the four most common DPYD polymorphisms associated with DPD deficiency has been implemented as routine screening clinical care in the Netherlands prior to start of treatment with capecitabine. In patients carrying one of these polymorphisms, dose adjustments are made according to the gene activity score.36,37 Unfortunately, we have no knowledge about the genotype of the patients in the fixed dose cohort because they were treated before the implementation of upfront genotyping. Prevalence of a partial DPD deficiency is around 3–5%. Therefore, we have to assume that a small group of patients in our cohort indeed had a partial DPD deficiency. The DPYD genotype is known for all the patients from the BSA-based dose cohort, and the mutant patients received a dose reduction; we do not think that this will influence our results. As all patients treated with a fixed dose in the mentioned time period were included in our analysis, we assume that the genetic distribution is comparable in the fixed-dose group of patients. However, these patients could not have received a dose reduction in the case of DPD deficiency and therefore this could lead to a small increase in toxicity risk in the fixed-dose group.
Besides toxicity, we also have investigated the effectiveness of given treatments. We hypothesized that if a fixed dose would (positively or negatively) influence the effectiveness of the treatment a survival difference should occur between the patients with a low and a high BSA value per treatment and indication. Of interest, we have found no statistical differences between BSA subgroups in PFS for CAPOX for mCRC or ECC/EOX for gastric cancer, nor for the DFS for CAPE + RT for laCRC. In addition, the observed PFS for CAPOX for mCRC and ECC/EOX for gastric cancer was comparable with the literature (8.6 and 24.6 months versus 8.0 and 19.2 months, respectively).38,39 Although we found no major differences in the effectiveness in all subgroups analyzed, not all regimens could be evaluated due to small sample sizes, and therefore we have to interpret these results with caution.
Our study has some limitations that need to be mentioned. Firstly, the retrospective nature of our data collection makes it difficult or even impossible to obtain toxicity and effectiveness data in a standardized fashion. However, we have evaluated combined toxicity scores, which consisted of severe capecitabine-specific toxicities or clinically relevant events due to toxicity. In general, these scores are well documented because of the large impact on the patient and treatment decisions. Moreover, patients were excluded when their patient file was not available or when visits were poorly documented. In the survival analysis there was a frequent loss to follow up and censoring of patients. Nevertheless, we found comparable survival probabilities for all subgroups as described in the literature. In addition, survival outcomes between different BSA groups were not different. However, the survival analysis was performed without a control group as in the BSA-based dose cohort survival data were not collected. Another limitation is the risk of confounding by hospital, treatment indication and dosing strategy. Although the fixed-dose cohort patients were included in a single hospital, we cannot argue that this might have influenced our results, because no major differences with the BSA-based dose cohort were identified.
Our study evaluated a large population of cancer patients treated with frequently used treatment regimens, thereby representing daily clinical care. In addition, we confirmed our results in another large and comparable cohort of patients. Although BSA-based dosing has been the standard choice in oncology for decades, there is little evidence for this approach. Therefore, there is a need for evidence that a fixed dose could be a well-tolerated alternative strategy for already existing anticancer agents such as capecitabine. Earlier, three small prospective studies demonstrated that fixed-dose capecitabine is feasible.40–42 However, these studies have not yet been translated into daily clinical care, probably because of their limited sample sizes. Sharma and colleagues showed that a fixed dose of capecitabine monotherapy in advanced colorectal cancer was well tolerated and effective in a cohort of 55 patients. 40 Also, capecitabine in a fixed dose as monotherapy and in combination with vinorelbine was shown to be well tolerated and effective in metastatic breast cancer patients.41,42 Additionally, a small retrospective study demonstrated that even a low fixed dose of 1000 mg twice daily for 14 days might have effectivity in metastatic breast cancer patients. 43 In a study by Rudek and colleagues, a large interpatient variability in capecitabine pharmacokinetics was shown without any influence of BSA, which also favors the fixed dosing strategy. 42 Furthermore, it would be interesting to investigate whether a fixed dosing strategy for capecitabine would lead to fewer prescribing mistakes and possibly to reducing costs, as has been demonstrated for some other drugs.18–20
In conclusion, we have shown that a fixed dose of capecitabine is as equally well tolerated as a BSA-based dose of capecitabine in several treatment regimens. Also, we have no data indicating that a fixed dose of capecitabine is less effective than a BSA-based dose. Our results indicate that fixed dosing of capecitabine is a reasonable and practical alternative for BSA-based dosing. Therefore, we would recommend implementing fixed dosing in future clinical studies and we have found no arguments why it could not be used in daily clinical care.
Supplemental Material
Supplementary_Information – Supplemental material for Comparison of toxicity and effectiveness between fixed-dose and body surface area-based dose capecitabine
Supplemental material, Supplementary_Information for Comparison of toxicity and effectiveness between fixed-dose and body surface area-based dose capecitabine by Femke M. de Man, G.D. Marijn Veerman, Esther Oomen-de Hoop, Maarten J. Deenen, Didier Meulendijks, Caroline M.P.W. Mandigers, Marcel Soesan, Jan H.M. Schellens, Esther van Meerten, Teun van Gelder and Ron H.J. Mathijssen in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
A part of this work was presented at the annual ESMO Congress (Madrid, Spain, September 2017, #527).
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.