
Abstract
The poor clinical outcome of hepatocellular carcinoma (HCC) patients is ascribed to the resistance of HCC cells to traditional treatments and tumor recurrence after curative therapies. Cancer stem cells (CSCs) have been identified as a small subset of cancer cells which have high capacity for self-renewal, differentiation and tumorigenesis. Recent advances in the field of liver CSCs (LCSCs) have enabled the identification of CSC surface markers and the isolation of CSC subpopulations from HCC cells. Given their central role in cancer initiation, metastasis, recurrence and therapeutic resistance, LCSCs constitute a therapeutic opportunity to achieve cure and prevent relapse of HCC. Thus, it is necessary to develop therapeutic strategies to selectively and efficiently target LCSCs. Small molecular inhibitors targeting the core stemness signaling pathways have been actively pursued and evaluated in preclinical and clinical studies. Other alternative therapeutic strategies include targeting LCSC surface markers, interrupting the CSC microenvironment, and altering the epigenetic state. In this review, we summarize the properties of CSCs in HCC and discuss novel therapeutic strategies that can be used to target LCSCs.
Introduction
Liver cancer is the fifth most common cancer globally and has an incidence of approximately 850,000 new cases per year. 1 Hepatocellular carcinoma (HCC) is the primary malignant neoplasm derived from hepatocytes and the dominant form of primary liver cancer, accounting for more than 80% of all liver cancer cases. 2 HCC often occurs in the presence of cirrhotic liver disease, arising from chronic infection of hepatitis B virus (HBV), hepatitis C virus (HCV) or alcohol abuse. 3 Increasing cases of HCC have also been observed to be accompanied by nonalcoholic fatty liver disease (NAFLD), which is a result of obesity and insulin resistance. 4 Different therapeutic approaches such as organ transplantation, surgical resection, transarterial chemoembolization (TACE), local radiofrequency ablation (RFA), and local microwave ablation are commonly adopted. 5 However, owing to the absence of pathognomonic symptoms, the majority of HCC cases are diagnosed at advanced stages for which efficient therapies are limited. 6 Sorafenib, a systemic targeted agent approved by the United States Food and Drug Administration (US FDA), is a first-line therapy with modest survival benefits and serves as an alternative in clinics for unresectable HCC cases. 7 Regorafenib and lenvatinib are two newly US FDA-approved medications for the treatment of HCC but have no superior effects to sorafenib.8,9
With a median survival following diagnosis of 6–20 months, HCC is ranked as the third leading cause of cancer death. 10 Long-term survival of HCC patients is hindered by high recurrence and drug resistance mainly due to the presence of liver cancer stem cells (LCSCs). 11 LCSCs are subpopulations of liver cancer cells which have high capacity for self-renewal, differentiation and tumorigenesis. Given the critical roles of LCSCs in tumor progression and therapeutic resistance, this article will discuss the characteristics of LCSCs and promising therapeutic strategies to target them.
LCSC origin
The origin of cancer stem cells (CSCs) from liver stem/progenitor cells is inferred from the fact that LCSCs share a substantial number of similar features with normal stem cells. Overall, 28–50% of HCC cells were reported to express progenitor cell markers such as CK7 and CK19, suggesting that at least a portion of HCC cells have intermediate characteristics between progenitors and differentiated mature hepatocytes. 12 The development of HCC is always associated with long-term inflammation that is induced by chronic HBV or HCV infection, alcoholic/NAFLD or chronic exposure to toxicity. 1 Such persistent inflammation leads to the expansion of stem/progenitor cells with accumulated epigenetic or genetic alterations. 13 Furthermore, the inflammatory microenvironment facilities the transformation of normal liver stem cells to LCSCs. 14
Reprogramming and dedifferentiation of non-CSCs have also been considered as a leading cause for the acquisition of CSC-like features in tumor cells. Mature hepatocytes, hepatoblasts and biliary cells can transform into LCSCs during liver injury/regeneration or under oncogenic dedifferentiation. 15 For instance, the loss of p53 drives the dedifferentiation of mature hepatocytes into progenitor-like cells and further leads to the development of HCC with gene mutations in Wnt and Notch signaling pathways. 16 CHD1L has also been reported as a potential development-related lineage oncogene that promotes the dedifferentiation of HCC cells through modulation of chromatin configuration at key transcription factors such as the estrogen-related receptor beta (ESRRB) gene and transcription factor (TCF) 4. 17 Interestingly, several studies have demonstrated that hepatocytes could be derived from bone marrow stem cells, which shed some light on transdifferentiation in liver cancer progression. 18
LCSC surface markers
CSCs have been identified in various tumor types, including breast cancer, lung cancer, ovarian cancer and prostate cancer. 19 The identification and isolation of CSCs mainly rely on the use of surface markers. During the past few decades, new developments have enabled us to identify putative specific surface markers for LCSCs, providing us with opportunities to explore potential biological functions, signaling pathways and therapeutic approaches. Epithelial cell adhesion molecules (EpCAM), CD133, CD90, CD13, CD44, OV-6, ALDH and K19 are widely recognized as LCSCs surface markers. Although their functions in LCSCs are not fully understood, studies have shown that they exert tremendous control over the acquisition of tumorigenesis, invasiveness, self-renewal, metastasis and drug resistance. We summarize molecules identified as specific surface markers of LCSCs in Table 1.
Surface markers, functions and clinical characteristics in LCSCs.

ABC, adenosine triphosphate binding cassette; AFP, alpha-fetoprotein; EMT, epithelial to mesenchymal transition; HBV, hepatitis B virus; LCSC, liver cancer stem cell; N/A, not applicable; ROS, reactive oxygen species; TGF, transforming growth factor.
EpCAM
EpCAM is a type I transmembrane glycoprotein, which is frequently expressed in multiple carcinomas. EpCAM not only functions in the process of cell–cell adhesion, but also participates in cell migration, metastasis, proliferation, cell cycle and cell differentiation. EpCAM serves as a biomarker of HCC and is especially highly expressed in HBV-positive patients. 33 It is also extensively demonstrated as a marker for stem/progenitor cells of adult liver cells. 34 A series of gene profiling and pathway analyses have indicated that the EpCAM+ alpha-fetoprotein+ (AFP+) HCC subtype has CSC-like characteristics, poor prognosis and shorter survival. 35 EpCAM+ HCC cells commonly possess CSC traits including the capacity for self-renewal, differentiation and tumorigenesis, and a signature of chemotherapy resistance.35,36 EpCAM is known to be a direct transcriptional target of Wnt-β-catenin signaling in HCC cells and has a significantly reduced expression pattern in response to Wnt-β-catenin signaling antagonists. 37
CD133
CD133 (human prominin-1, PROM1) is abundantly expressed in both the cytoplasm and nucleus of HCC tumor tissues compared with adjacent normal liver tissues. 38 The expression of CD133 was a significant prognostic marker for the overall survival of HCC patients, 39 and HCC containing CD133+AFP+ cells can be further classified as a poor prognostic subgroup. 40 CD133+ cells display typical CSC-like abilities such as spheroid formation, chemoresistance, migration and tumorigenesis. Mechanistically, CD133 may facilitate CSC characteristics via stabilizing EGFR-Akt signaling or regulating neurotensin/interleukin (IL)-8/CXCL1 signaling.41,22
CD13
CD13 (aminopeptidase N) is a membranous glycoprotein, which is closely linked with tumor progression. CD13+ cells form cellular clusters mainly in cancer foci and are predominated in the G0 phase of the cell cycle. 24 The high expression of CD13 in HCC patients is a significant risk factor for early recurrence, poor prognosis and shorter survival. 42 Inhibition of CD13 can block the tumor-initiating and self-renewal ability of LCSCs. Proliferation results have further demonstrated that CD13+ HCC cells are resistant to chemotherapy. Mechanistically, CD13 may reduce reactive oxygen species-induced DNA damage after chemotherapy or radiotherapy to prevent HCC cells from apoptosis. 24
CD44
CD44 is a cell surface glycoprotein, which participates in multiple cellular processes including cell growth, survival, differentiation and motility. CD44 is preferentially expressed in CD133+ populations and contributes to the CSC-like features synergistically. CD133+CD44+ HCC cells have higher clonogenicity, whereas the CD133+CD44– subset is with attenuated phenotype. 43 CD90+44+ HCC cells have been shown to be more aggressive than their CD90+CD44– counterpart and form metastatic lesions in the lung. 44 Thus, CD44 is commonly used to distinguish CSC subpopulations in combination with other surface markers.
The role of CSCs in the liver
The properties of stem cells appear to descript CSCs as a small subset of cancer cells which may contribute to the diversity and heterogeneity of tumors. 45 CSCs are widely considered to be more tumorigenic than other nonstem cancer cells and resistant to multiple anticancer therapies, including chemotherapy and radiotherapy. 45 Here we summarize the role of CSCs in the liver, particularly focusing on their functions in tumor initiation and growth, metastasis, therapeutic resistance and recurrence.
LCSCs and tumor initiation
A growing body of evidence has shown that CSCs play essential roles in liver tumor initiation. The isolated LCSCs that are featured with CSC surface marker expression profiles have high tumorigenic capacity. For example, CD133-positive Huh7 and PLC8024 cells were shown to possess higher tumorigenic and proliferative potential and have lower expression of mature hepatocyte markers than CD133– counterparts. 46 It should be noted that CD133– HCC cells can give rise to CD133+ cells in subcultures. 47 However, CD133 does not appear to be critical in tumor initiation or development, as both CD133+ and CD133– cells derived from colon cancer are capable of initiating tumors in mice. 48 Since some CSC markers can be detected in lymphocytes, the isolation of CD45− cells was used to define nonlymphatic cells in tumor tissues. Yang and colleagues demonstrated that CD90+ cells derived from HCC cell lines displayed increased tumorigencity. 49 Moreover, the blood samples that were separated from liver cancer patients by virtue of CD45–CD90+ expression could efficiently generate tumor nodules in xenograft models. 49 A recent study illustrated that a highly tumorigenic HCC cell line, Dt81Hepa1-6, which was derived from Hepa 1-6 HCC cells through in vivo passage, were characterized by EpCAM+ expression. 50 Sun and colleagues showed that as little as 300 EpCAM+CD45– cells isolated from HCC patient samples could initiate tumors in NOD/SCID mice, whereas 1 × 104 EpCAM-CD45– cells failed to form tumors, suggesting that HCC cells with stem/progenitor cell traits are much more likely to form tumor in vivo. 51
The mechanisms underlying the high tumorigenicity of LCSCs are not fully understood. This phenomenon could be partially explained by the fact that representative genes involved in cancer stemness such as CSC surface markers participate in the activation of core signaling pathways in cancer. For instance, the activation of Wnt/β-catenin pathway is a major feature for the isolated EpCAM+ HCC cells and functions in tumor initiation. 35 Akita and colleagues showed that the expression of human embryonic stem cells (hESCs) marker c-Myc at low levels in HCC cells induced the activation of reprogramming transcription factors and the expression of CSC markers including Nanog, EpCAM and Oct4. 52 c-Myc is a potent oncogene through the induction of telomerase and the dysregulation of microRNA (miRNA). 53 Thus, the activation of c-Myc could drive the carcinogenesis linked with cancer stemness. Moreover, we have also noticed that the expression of multiple LCSCs surface markers are not completely overlapping. EpCAM+ and EpCAM– HCC cells which initiate tumors in different sizes in vivo were found to equally express CD133, suggesting that LCSC populations with different surface marker expression patterns were characterized by heterogeneous signaling networks. 54
LCSCs and therapeutic resistance
The effectiveness of standard anticancer therapies such as chemotherapy, sorafenib and radiotherapy are always impaired by CSC-mediated resistance. It has been well recognized that enriched LCSCs from HCC cells are commonly resistant to multiple treatments.
Sorafenib is an oral multikinase inhibitor and has become the first-line treatment in patients with advanced HCC. Sorafenib targets cell surface tyrosine kinase receptors such as vascular endothelial growth factor receptor, platelet-derived growth factor receptor and epidermal growth factor receptor (EGFR) as well as serine/tyrosine kinases including Raf, FMS-like tyrosine kinase-3 (Flt-3) and c-kit. 55 An in vitro study demonstrated that sorafenib could efficiently reduce cell viability and induce apoptosis in HCC cell lines. 7 However, advanced HCC patients only had a survival benefit of 3 months after sorafenib monotherapy. 7 The application of sorafenib has been hampered due to drug resistance. In addition, long-term treatment with sorafenib can lead to a more aggressive phenotype since cancer cells undergo epithelial to mesenchymal transition (EMT), which is closely associated with the function of CSCs. 56 Sorafenib can upregulate stemness genes Nanog, Sox2 and Oct4 in EpCAM-positive HCC cells and exacerbate disease progression. 57 Enriched proportions of CD44+ and CD44+CD133+ HCC cells were also observed in sorafenib-resistant cells, suggesting that treatment with sorafenib could promote cancer stemness in HCC. 56 Interestingly, LCSCs derived from HCC cell lines were found to be relatively resistant to sorafenib and manifested with improved viability, reduced apoptosis and stem cell differentiation gene expression profiles. 58 These results highlight the role of sorafenib treatment in LCSC maintenance as well as the presence of LCSC-mediated sorafenib resistance.
The efficiency of chemotherapeutic agents on LCSCs has also been evaluated. Chemotherapies could increase the CSC population in HCC cells. For example, Ma and colleagues reported that doxorubicin and 5-FU treatment to unsorted HCC cells or cells derived from CD133+ Huh7-induced xenograft tumors significantly enriched the CD133+ subpopulation, whereas the proportion of CD133+ cells in untreated cohorts remained relatively unchanged. Moreover, CD133+ HCC cells conferred resistance to doxorubicin and 5-FU. 59 CD13 expression was reported to increase significantly towards doxorubicin or 5-FU treatment in HCC cells. The isolated CD13+CD133+ HCC cells were more resistant to doxorubicin in comparison with CD13–CD133+ and CD13–CD133– cells. 24 The reported correlations between LCSCs and other therapeutic resistance are summarized in Table 2.
Reported therapeutic resistance in LCSCs.
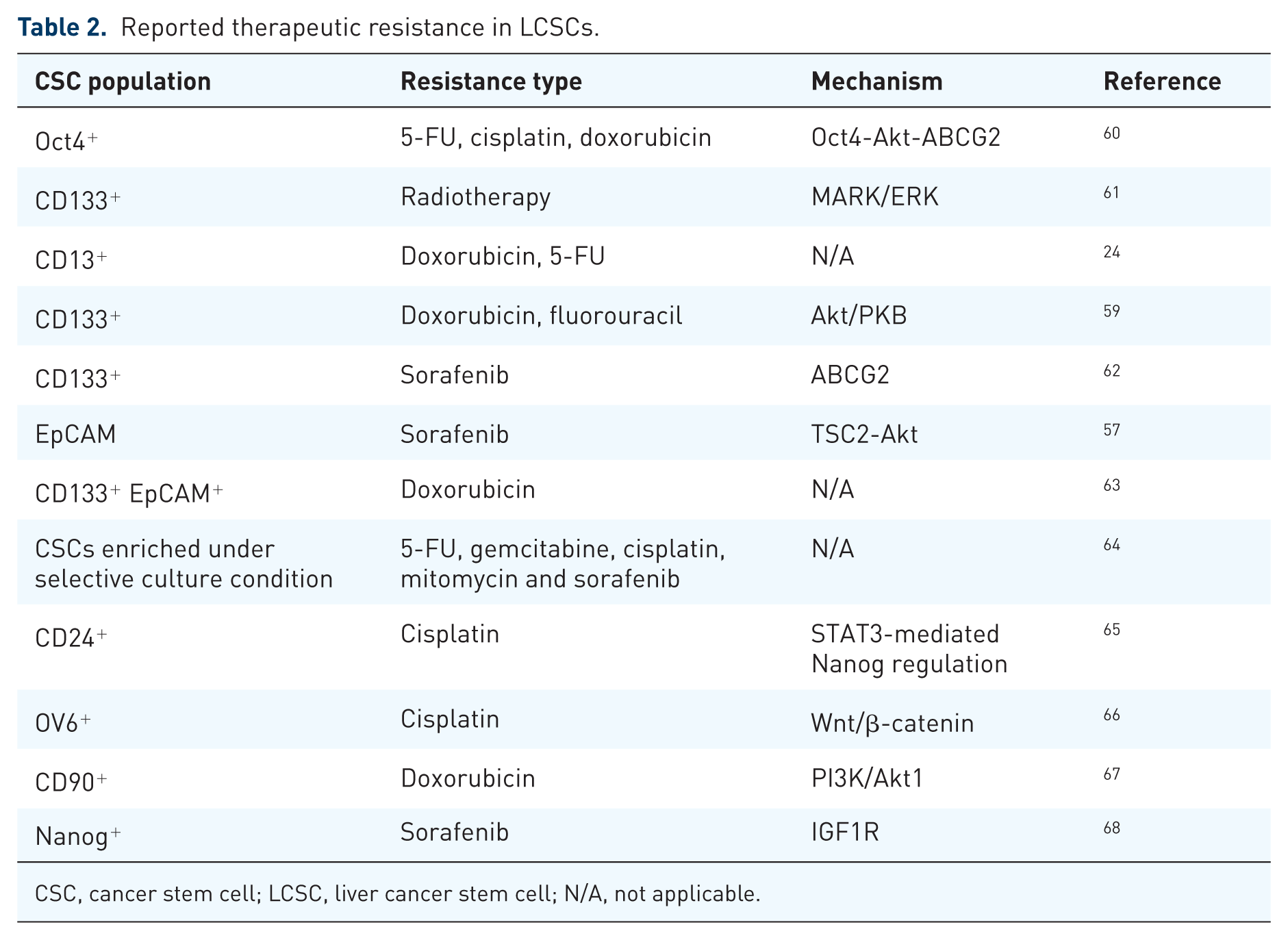
CSC, cancer stem cell; LCSC, liver cancer stem cell; N/A, not applicable.
The adenosine triphosphate (ATP) binding cassette (ABC) transporters are a family of membrane transporter proteins that have a major impact on drug efflux and are primarily responsible for drug resistance. 69 Expression of LCSC surface markers is frequently associated with the upregulation of ABC transporters. Zhu and colleagues reported that CD133+CD44+ HCC cells were more resistant to chemotherapeutic agents due to the enhanced expression of ABC transporters (ABCB1, ABCC1 and ABCG2). 43 ABCG2 and Oct4 were overexpressed in enriched CD90+CD133+ LCSCs, which to a large extent contributed to the chemoresistance. 70 Wang and colleagues showed that Oct4 overexpression led to the activation of TCL1, Akt and ABCG2 and exhibited a chemoresistant phenotype. This study further indicated that a direct pathway of Oct4-TCL1-Akt-ABCG2 or a combination of Oct4-TCL1-Akt with Akt-ABCG2 pathway could mediate chemoresistance in HCC. 60
In addition to the involvement of ABC transporters, other mechanisms have also been introduced to explain the critical role of LCSCs in anti-liver cancer therapy. For example, EpCAM+ subset in HCC was found to abundantly express the chromatin remodeling enzyme CHD4. The activation of CHD4 could collaborate with PARP and repair double strand breaks to enhance chemoresistance to DNA damage reagents in HCC. 71 Activation of cell survival response was also proposed to explain LCSC-mediated resistance. The isolated CD133+ LCSCs conferred resistance to 5-FU and doxorubicin through the activation of the Akt/PKB and Bcl-2 survival pathway. 59 In another study, it was reported that the acquired sorafenib resistance was mediated by the activation of TSC2-Akt cascades, which could contribute to the upregulation of cancer stemness and enhance tumorigenicity. 57
LCSCs and metastasis
Accumulating evidence has demonstrated that CSCs are a primary cause of metastasis for their roles in new tumor initiation at local or distant sites. As a multistep process, the metastatic cascade requires the invasion of primary tumor cells into adjacent tissues, the entry of tumor cells into circulatory systems through the extracellular matrix, the extravasation through vascular walls into distal tissues and the proliferation in competent organs. 72 Yamashita and colleagues demonstrated that EpCAM+AFP+ HCC tumor cells were abundantly located at the invasive front, suggesting that CSC marker expression is correlated with HCC invasion and metastasis. 35 It also has been documented that tumorigenic CD90+CD45– cells were detected in the circulation of liver cancer patients, but not in disease-free controls or patients with cirrhosis. 73
EMT is considered to be a key process that drives tumor cell metastasis. During EMT, the epithelial cells lose their differentiated abilities and acquire mesenchymal properties. 74 Recent studies have shown that the biological functions of LCSCs are closely related to the EMT phenotype. The coexpression of EMT-associated genes and CSC markers has been frequently identified in HCC. For instance, K19+ HCC cells displayed CSC-like features together with the high expression of EMT markers. 31 By examining 48 HCC patients who underwent surgical resection, CD44 expression levels were found to be associated with tumor metastasis and recurrence. 75 Mima and colleagues reported that the effect of CD44 on metastasis was due to its role in transforming growth factor-beta (TGF-β)-mediated EMT, leading to decreased cell–cell adhesions and enhanced capacity to migrate. 76 CD44+ HCC cells had high expression levels of EMT markers N-cadherin and vimentin. 77 Moreover, the knockdown of CD44 resulted in EMT reversion and decreased lung metastasis by repressing ERK/Snail signaling. 77 In another study, the altered low expression of CD133 reversed the EMT phenotype and attenuated the self-renewal of LCSCs. 23
In fact, the core signaling pathways that govern the EMT and cancer stemness are considered to be intricately linked. For example, as a crucial regulator of EMT, the upregulation of Slug has been shown to increase the percentage of CD133+ subpopulation among HepG2 cells and induce stronger cancer stemness properties. 78 The ectopic expression of Twist2 increased the invasiveness and metastasis of human cancer via the downregulation of E-cadherin. 79 Liu and colleagues reported that the exogenous overexpression of Twist2 could enhance the expression of CSC-related genes including Bmi-1, Sox2, CD24 and Nanog, and augment the self-renewal capacity through the transcriptional activation of CD24. 80 TGF-β1 is well known as an EMT inducer and reported to increase the expression of CD44 in HCC. 81 Park and colleagues demonstrated that CD44 and TGF-β1 could synergistically promote the CSC properties and EMT phenotype through the Akt/GSK-3β/β-catenin pathway in HCC cells, leading to a more aggressive HCC progression. 82 Study has shown that K19 is highly expressed in invasive and metastatic HCC and could serve as a putative CSC marker. K19+ HCC cells were found to have EMT gene expression profiles and mesenchymal characteristics. 31 Such an EMT phenotype is dependent on the activation of TGF-β/Smad signaling, and could be eliminated by K19 knockdown or TGF-β R1 inhibitors. 31
LCSCs and recurrence
The high tumor recurrence rates after curative treatments impair long-term survival of HCC patients. HCC recurrence may be ascribed to LCSCs from several perspectives. Firstly, it is proposed that exposure to standard therapies largely reduces the nonstem cancer cells but has limited therapeutic effects on LCSCs. LCSCs display intrinsic resistance to chemotherapy and radiotherapy, which in turn results in the survival of a population of tumorigenic cancer cells. 73 Secondly, EMT could confer LCSCs with more aggressive traits and help these cells to survive standard anticancer therapies, subsequently being at least partly responsible for disease recurrence. The existence of EMT and LCSCs has been identified to account for tumor recurrence in HCC patients who were treated with RFA. 83 It also has been reported that β-catenin accumulation in HCC is associated with incidence of recurrence after liver transplantation. This study suggested that β-catenin signaling activation causes normal HCC cancer cells to differentiate into malignant, immature hepatocyte progenitors and serves as a basis for HCC recurrence. 84 Moreover, EpCAM+ circulating tumor cells which displayed both CSC and EMT phenotypes were more likely to undergo tumor recurrence after surgical resections. 51 Another perspective on liver cancer recurrence is related to the interaction between LCSCs and the microenvironment. The LCSC microenvironment comprises a diversity of cytokines (TGF-β), growth factors and cellular elements. The elevated level of IL-6 in the LCSC niche of HCC was associated with aggressive metastasis and recurrence. 85 In addition, the activation of multiple signaling cascades such as the TGF-β pathway in the tumor microenvironment could reciprocally promote cancer stemness and drive transformation of normal liver stem cells to LCSCs. 86 In liver cancer, CD133+ LCSCs were reported to have higher levels of proangiogenic factor vascular endothelial growth factor (VEGF). 87 Further study demonstrated that HCC patients with early recurrence displayed enhanced Nanog expression and VEGFR2 activation among CD133+ HCC cells. RFA-induced VEGF could increase the proportion of CD133+ CSCs and enhance cancer stemness by inducing the expression of Nanog. 88
Therapeutic strategies of LCSCs
A recent study has shown that sorafenib treatment significantly reduces the CD90+ subpopulations with attenuated c-Kit phosphorylation but leads to the enrichment of EpCAM+ cells. 89 In vivo study further demonstrated that sorafenib could completely inhibit lung metastasis mediated by CD90+ LCSCs, but failed to suppress the primary tumor growth induced by EpCAM+ CSCs. This therapeutic effect can be at least partially explained by the fact that sorafenib suppresses the production of extracellular vesicle which containing TGF-β mRNA in CD90+ cells. 89 Acyclic retinoid (ACR) is a synthetic vitamin A-like compound and has been found to prevent the recurrence of HCC. ACR has been shown to suppress the expression of a Myc family member, MYCN, which is tightly involved in both CSC and Wnt/β-catenin signaling. ACR is more sensitive to EpCAM+ cells, which are featured with MYCN expression, and serves as a potential therapeutic agent to target LCSCs. 90
Although there have been therapeutic advances in the field of LCSCs, treatment options and therapeutic effects remain limited. Further investigations to develop novel anti-LCSC therapies are yet to be conducted. It is well recognized that Wnt/β-catenin, Notch, Hedgehog, and TGF-β pathways are inherent signaling pathways to regulate self-renewal, proliferation and differentiation of normal stem cells. These cellular signaling pathways have been identified to be dysregulated during hepatocarcinogenesis and crucial determinants of stem cell properties as well as tumorigenesis. Thus, targeting these signaling pathways provides a promising strategy for cancer therapy. Other therapeutic strategies have also been proposed with the specific aim of disrupting the LCSC microenvironment, eliminating LCSC surface marker expression, alternating epigenetic status, modulating autophagy and inhibiting ABC transporters.
Targeting the Wnt/β-catenin signaling pathway
Wnt/β-catenin signaling is evolutionarily conserved in embryonic development and normal tissue homeostasis. 91 The canonical Wnt/β-catenin pathway is suggested to play a major role in HCC progression, while the non-canonical Wnt/β-catenin signaling is less detailed. 92 In the absence of Wnt signaling, the cell membrane pool of β-catenin is bound to E-cadherin. The cytosolic pool of β-catenin is kept at low levels through continuous phosphorylation by kinase glycogen synthase CK1 and GSK3β and a multiprotein complex that contains adenomatous polyposis coli (APC), Axin and WTX. The canonical Wnt/β-catenin cascade is activated by the engagement of Wnt ligands with Frizzled receptors, leading to the accumulation of cytosolic β-catenin. Then, the β-catenin translocates into the nucleus and interacts with TCF/LEF factors to induce the transcriptional activities of Wnt signaling target genes such as cyclin D1, c-Myc and Survivin. 93
Hyperactivation of the Wnt/β-catenin pathway has been observed in at least one third of HCC cases. The accumulation of cellular and nuclear β-catenin, which is a hallmark of canonical Wnt pathway activation, has been found in 33–67% HCC. 94 Wnt/β-catenin signaling is responsible for maintaining the self-renewal capacity and inhibiting the differentiation of LCSCs. ALDH1 and CD133 double-positive HCC tissue samples had elevated expression levels of Wnt1 and β-catenin. 95 The inhibition of CBP/β-catenin signaling was found to result in the reduced expression of CD133 in HCC cells. 96
Small molecular agents have been studied extensively to target Wnt/β-catenin signaling and negatively modulate the cancer stemness in liver cancer. However, currently there is no available US FDA-approved drug for human use. CWP232228 was first patented as a potent small molecular inhibitor which antagonizes the binding interaction between β-catenin and TCF in the nucleus. This compound has been shown to preferentially inhibit breast CSC subpopulations rather than normal tumor cells and could safely suppress tumor formation and metastasis in breast cancer. 97 The therapeutic effect of CWP232228 has also been confirmed in LCSCs. The compound could inhibit the basal expression of Wnt1 and TCF4 in a dose-dependent manner and subsequently downregulate β-catenin-responsive genes and suppress the tumor sphere formation. 95 Another small molecular inhibitor, FH535, was designed as an antagonist to PPARγ and PPARδ, inhibiting the recruitment of GRIP1 to β-catenin. 98 It has been uncovered that FH535 inhibited transcriptional activities of β-catenin target genes in LCSCs. 92 FH535 suppressed the proliferation of LCSCs and parental HCC cells, suggesting that FH535 is a potent anticancer therapy in the liver. 92 Moreover, the combination of FH535 and sorafenib was reported to disrupt the bioenergetics of HCC cells by targeting mitochondrial respiration as well as glycolytic flux and synergistically inhibiting HCC/LCSC proliferation.94,99 Matrine is the major active ingredient of Sophora flavescens and shows efficient therapeutic effects on various cancer types. However, its low bioactivity impeded its implication in clinics. 100 WM130 is a novel derivative of matrine with antitumor effects and improved pharmacological activities. Studies have revealed that WM130 could decrease phosphorylation of GSK3β (Ser9), resulting in degradation of β-catenin. 101 WM130 was found to suppress the primary and subsequent HCC spheres, inhibit the proliferation of doxorubicin-resistant hepatoma cells and reduce the expression of EpCAM. 102 A recent study identified MASM as another derivative of matrine to treat LCSCs. MASM greatly reduced cancer stemness and promoted the expression levels of mature hepatocyte markers in the enriched Hep3B and Huh7 spheroids, suggesting it as a promising candidate in HCC treatment. 103
OMP-54F28 is a fusion protein that is composed of the cysteine-rich domain of Frizzled family receptor 8 (Fzd8) receptor fused to the immunoglobulin (Ig)G1 Fc regions and was able to compete with the Fzd8 receptor for its ligands to suppress the Wnt/β-catenin signaling. 104 Preclinical studies have shown that OMP-54F28 could reduce tumor growth and decrease CSC frequency alone or in combination with other agents such as gemcitabine. 104 Gemcitabine has emerged as the first-line treatment of multiple cancer types. However, tumor cells develop gemcitabine resistance rapidly. The proportion of CD44+ cells increased from 12.7% to 13.9% when treated with gemcitabine alone in pancreatic tumor cells. The combined administration of OMP-54F28 and gemcitabine led to only 1.9% of CD44+ cells, suggesting that OMP-54F28 could facilitate the therapeutic effects of gemcitabine. 105 A phase Ib clinical trial (ClinicalTrials.gov identifier: NCT02069145) looking at the combination of OMP-54F28 and sorafenib to evaluate the safety and pharmacokinetics/pharmacodynamics in HCC patients was completed in 2017. 106 Although currently no results have been posted for this clinical study, a dose escalation study in solid tumors (ClinicalTrials.gov identifier: NCT01608867) has indicated that OMP-54F28 is well-tolerated at two times the target efficacious dose, providing valuable information for its safety on clinical implications. 107
Targeting Notch signaling pathway
The Notch signaling pathway is essentially involved in several fundamental cellular processes including proliferation, survival, apoptosis, differentiation and cell adhesion. 108 Notch signaling contributes to self-renewal and differentiation of CSCs in many cancer types. In liver cancer, Notch-specific gene expression signature is harbored in 30–35% human HCC samples and is associated with the expression of Sox9, which marks the pluripotent population in liver. 109 Increased expression of Notch1 has been shown in CD90+ HCC cells and is responsible for mediating self-renewal, invasion and migration. 110 The loss of Notch1 exhibited a decreased expression of HES1 and cyclin E, whereas the expression of p53, p21 and p27 was significantly induced. 111 Zhu and colleagues evidenced that Notch2 was activated and sustained the cancer stemness in LCSCs. The activation of Notch2 was positively associated with poor clinical outcomes of HCC patients. 112 Moreover, Notch3 has been identified as a positive regulator in the cancer stemness and to regulate the differentiation, chemosensitivity and survivals of hepatoma cells. 113 The importance of Notch family members in CSCs regulation is further supported by the fact that Notch signaling activated a subpopulation of Sox9 and K-19 positive progenitors in liver oncogenesis process. 114 A complicated cross-talk between Notch and Wnt/β-catenin signaling has also been proposed. Wang and colleagues have indicated that the expression of Notch1 intracellular domain (NICD) 1 is dependent on the activation of Wnt/β-catenin signaling pathway, suggesting that Notch1 is downstream of Wnt signaling. 115 In addition, a nonproteasome-mediated feedback loop between Notch1 and Wnt/β-catenin signaling was observed in LCSCs. 115 Oppositely, another study suggested that the expression levels between Notch3 and β-catenin were inversely correlated. 113 Overall, the outcome of the intricate interaction between Wnt and Notch signaling pathways is still debatable.
Notch signaling is activated through the binding interaction between transmembrane Notch receptors (Notch1–4) and Notch ligands (Jagged 1,2 and Delta-like 1,3,4). 108 This interaction induces proteolytic cleavage of Notch receptor by γ-secretase and results in the release of NICD translocate to the nucleus where NICD binds with the DNA-binding transcription factor CSL/RBP-Jκ and recruits MAML1 as a coactivator to form a transcriptional activation complex. 116 The well-recognized target genes of the Notch signaling cascade are HES/HEY families. 116
γ-secretase inhibitors are a class of Notch inhibitors that have been extensively explored. Several γ-secretase inhibitors have shown antitumor effects and undergo clinical trials in breast, pancreatic, colorectal cancer and renal cell carcinoma. 116 To date, no γ-secretase inhibitors have been processed to clinical trials in liver cancer. In vitro study demonstrated that γ-secretase inhibitors decreased the proliferation, blocked cell cycle and induced apoptosis in HCC cells. 117 Moreover, γ-secretase suppressed the maturation of HCV core proteins, which is involved in the processing of HCV propagation and pathogenesis. 118 PF-03084014, a γ-secretase inhibitor, has been identified as a novel Notch inhibitor by blocking Notch cascades and shown to have antitumor effect on multiple cancer types. 119 It is currently under phase I or phase II clinical trials for the treatment of breast cancer, lymphoma and aggressive fibromatosis. In liver cancer, it could impair the self-renewal and proliferation of LCSCs, resulting in reduced tumor growth and metastasis. 120 Such inhibition effects on cancer stemness are achieved by suppressing Notch1 signaling and STAT3 activity. 120
Other alternative strategies such as decoys, blocking peptides and antibodies against Notch receptor or ligands are also considered as Notch signaling-targeted therapies. 114 The interactions between Notch ligands and receptors are essential for Notch signaling initiation. Monoclonal antibodies such as NRR1, NRR2, NRR3 monoclonal antibodies can specifically target to Notch regulatory region and recognize Notch family receptors, resulting in the blockage of Notch signaling. 114 This interference effects could also be accomplished by other antibodies, which compete with endogenous ligands including Jag1, Dll1and Dll4. 114 Decoys are soluble form of Notch receptors or ligands extracellular regions which mimic and compete with endogenous proteins. 121 These Notch inhibitors have lower gastrointestinal toxicity compared with γ-secretase inhibitors and have strong potential for further clinical translation. 121 In the nucleus, the released NICD binds with DNA-binding protein CSL and recruits MAML1 as coactivator, resulting in the transcription of Notch target genes. Blocking peptides such as DN-MAML1, SAHM1 reduce the formation of NICD/CSL protein complex and subsequently suppress the transcriptional activation of target genes.122,123
Targeting the Hedgehog signaling pathway
The Hedgehog (Hh) signaling pathway is implicated in a wide variety of cellular process during embryonic development and adult tissue homeostasis. The Hh cascade is dysregulated in various cancer types including pancreatic, breast and prostate cancer. 124 The aberrant activation of the Hh signaling pathway contributes to the maintenance of stemness in CSCs and the acquisition of EMT. 125 In liver cancer, the activation of the Hh pathway can promote metastasis and invasion, whereas the decreased activity of Hh signaling inhibits HBx-induced cell migration, anchorage-independent growth and tumor development in HCC. 126 It has been shown that the expression of GLI-1 and GLI-2 is significantly increased in HCC patients.127,128 The silencing of GLI-1 led to increased expression of E-cadherin and decreased expression of Snail and vimentin in HCC cells. 129 Smoothened (SMO) was abundantly expressed in highly tumorigenic CD133+ HCC cells, providing a link between Hh signaling and its role in liver cancer stemness maintenance.130,131 Recently, Ding and colleagues reported that an aberrant Hh signaling pathway was activated in a specific subpopulation of Huh7 cells which is featured with CD133–/EpCAM– expression profiles. 132 Such an Huh7 subpopulation showed increased expression of Hh transcriptional factor GLI-2 and ABCC1 genes and exhibited strong resistance to sorafenib. 132 This study also suggested that GLI-2 had a negative impact on the sensitivity of hepatoma cells to sorafenib through the ABCC1 transporter. 132
The components of the Hh signaling pathway are Hh ligands (Sonic Hh, Indian Hh and Desert Hh), the transmembrane receptor Patched (Ptc), the signal transducer SMO and GLI family zinc finger transcriptional factors. 133 Targeting the Hh signaling pathway has preferentially focused on the antagonists of SMO and GLI1. Cyclopamine was firstly used as an efficient Hh inhibiter via binding to SMO directly. 134 However, it has been reported that cyclopamine fails to reduce the cell viability of Hep3B. 135 KAAD-cyclopamine, which targets oncogenically mutated SMO, suppresses Hh signaling activity by 50% and reduces the expression of the hepatocarcinogenic oncogene c-Myc in Hep3B cells. 135 GDC-0449 is a small molecular inhibitor that binds to the SMO receptor to suppress Hh signaling pathway. 136 This inhibitor is under active investigation in clinical trials for the treatment of primary or recurrent cancers. In vitro study indicated that GDC-0449 efficiently abrogated the effects of Hh signaling within liver parenchyma, HCC nodules and reduced liver fibrosis. 137 GDC-0449 also decreased the CD44 positive cells in primary liver tumors, suggesting that GDC-0449 could reduce the subpopulation of liver cancer cells with cancer stemness features. 137 As another SMO antagonist, LED225 could attenuate hepatic inflammation in mice with NAFLD and decrease the expression levels of proinflammatory cytokines such as TNF-α, IL-1β, MCP1, and IL-6 via suppressing the Hh pathway. 138 Since that NAFLD is a significant risk factor for developing liver cancer, the application of LED225 is a promising therapeutic agent for the treatment of liver cancer. LED225 is currently under phase I clinical trial to test its safety and maximum safe dose in patients with advanced or metastatic HCC and Child–Pugh A/B7 cirrhosis (ClinicalTrials.gov Identifier: NCT02151864). GANT61 is a GLI antagonist and displays antitumor effects in several cancer types. 139 GANT61 has been reported to inhibit cell viability, spheroid formation and GLI-DNA binding in pancreatic cancer. 140 GANTA61 also inhibited the pluripotency maintaining factors such as Nanog, Oct4, Sox2 and c-Myc in pancreatic CSCs. 140 Recently, study has been elucidated that GANT61 induces autophagy and inhibits tumor formation in HCC, suggesting it as a potent therapeutic agent for HCC. 141
Targeting the TGF-β signaling pathway
The TGF-β family members have been considered as crucial regulators in the self-renewal and differentiation of stem cells. 142 Multiple lines of evidences have been demonstrated that TGF-β plays bipartite roles in the development of liver cancer, functioning as a tumor suppressor at the early stages of liver diseases, whereas it acts as a tumor promoter to facilitate invasive and metastatic behaviors during cancer progression.143,144 The downregulation of TGF-β was shown to sensitize HCC cells to sorafenib treatment. 145 Tumor-associated macrophages (TAMs) secreted TGF-β1 and contributed to LCSC characteristics in HCC via inducing EMT. 146 It has been reported that TGF-β1 inhibited the expression of DNA methyltransferases DNMT1 and DNMT3, leading to the demethylation of CD133 promoter in CD133− Huh7 cells. 147 It has also been shown that IL6/STAT3 inhibitors could effectively eradicate LCSC features in HCC via indirectly disrupting the TGF-β pathway. 148
A number of TGF-β inhibitors have been in development as potential candidates for the treatment of LCSCs. The TGF-β ligands bind to TGF-β receptor (TβRI/TβRII) and form a heterodimer complex. 149 The complex recruits and phosphorylates the intracellular proteins SMAD2 and SMAD3, translocating to the nucleus and subsequently inducing the transcriptional activities of several target genes.149,150 One of the most frequently investigated TGF-β inhibitors is galunisertib (LY2157299), which suppresses the activity of TGF-β receptor I (TGFβRI). 151 Galunisertib reduced the expression of proliferative marker Ki67 and increased the apoptotic marker caspase-3 in HCC. 152 Moreover, this compound potentiated the therapeutic effects of sorafenib by inhibiting proliferation and inducing apoptosis. 152 Galunisertib was also found to inhibit migration and cell growth in HCC cells. 153 Although no study has been explicated for its anti-CSC activity in HCC, the inhibitory effects of galunisertib on CSCs have been shown in several other cancer types. For instance, galunisertib reduced the CD44high/PROCR+ population in breast cancer and prevented the development of paclitaxel-resistance CSCs. 154 Several phase I/II clinical trials are ongoing to test galunisertib alone or in combination with other drugs in HCC, suggesting it as a promising agent for HCC treatment. SB-505124 and SB-431542 are small molecule inhibitors of TGF-β receptor. They repressed the phosphorylation of SMAD2/SMAD3 and suppressed tumor cell growth and EMT in HCC.155,156 SB-505124 was found to decrease the CD44 expression in HCC cell lines. 157 SB-431542 was shown to reduce the subpopulation of CD90+, EpCAM+, CD133+ cells and decrease the expression of pluripotency factors including Nanog, Oct4 and Sox2 in cyclin D1-positive spheres in liver cancer. 158
Targeting the LCSC microenvironment
The functions of LCSCs are tightly regulated by cells in the tumor microenvironment. Liu and colleagues reported that single-cell-cloned LCSC derived from human liver cancer microvascular endothelial cells, was characterized with self-renewal and tumorigenic capacities. 159 The mimics of different carcinoma microenvironment induced the expression of specific tumor cell markers and had great impacts on the differentiation directions of the LCSC. 159
The tumor microenvironments are composed of stromal cells, immune cells, extracellular matrix, secreted cytokines and growth factors. 160 Such a microenvironment plays a critical role in the maintenance of CSCs through regulating the self-renewal or differentiation pathways, such as Wnt/β-catenin, Notch, Hh pathways and etc. 161 Cancer-associated fibroblasts (CAFs) are a major component of tumor stromal cells. 162 It has been well established that CAFs promote tumor growth and maintain the CSC features via paracrine activation. 163 In HCC, CAFs derived from hepatic tissues have strong anchorage-independent capability and clonogenicity. 164 More than 50% of these CAFs were shown as CD90+/CD44+. 164 CAFs isolated from HCC clinical specimens promoted the cancer stemness through c-Met/FRA1/HEY1 signaling mediated by HGF. 165 TAMs are another class of tumor stroma and serve as cancer stemness promoters. Wan and colleagues have demonstrated that the incubation of CD44+ HCC cells with TAMs helped the expansion of CD44+ cells as well as their sphere formation and tumorigenesis. IL-6 produced by TAMs in HCC could activate the STAT3 pathway to promote HCC stem cells. 166
The hypoxic nature of the tumor microenvironment contributes to the increased LCSC proportions in HCC cells. 167 The elevated expression of HIF-1α and lower reactive oxygen species activities were found in the induced LCSCs. 167 The mechanism underlying hypoxia-induced CSC expansion has been implicated extensively. As a hypoxia responsive gene, artemin (ARTN) enhanced the tumor sphere formation of HCC cells and increased the CD133+ CSC population. 168 SUMO protease 1 (SENP1) has been reported to promote hypoxia-induced cancer stemness through enhancing HIF-1α deSUMOylation and increasing the stability and transcriptional activity of HIF-1α. 169 The destruction of the hypoxic microenvironment is a rationale strategy to develop potential therapeutic agents targeting LCSCs. Wang and colleagues developed a polylactic-co-glycolic acid (PLGA)-encapsulated disulfiram (DS) whose encapsulation efficiency, drug-loading content and controlled release were satisfied with the therapeutic needs. 170 The combination of DS-PLGA and copper significantly reduced hypoxia-induced CSCs and abolished the sphere-forming abilities of HCC cells. 170 This nanomedicine also performed anti-HCC efficacy in mouse xenograft model and improved the cytotoxicity of 5-FU and sorafenib. 170 Since DS and PLGA are US FDA-approved clinical used products and commercially available, the development of DS-PLGA is a promising anticancer strategy.
Targeting LCSC surface markers
Several therapeutic agents targeting CSC surface markers directly have been developed. Oncolytic measles viruses (MV) termed MV-141.7 and MV-AC133 were reported to specifically target CD133 in HCC and selectively lysed CD133+ tumor cells. 171 The infection of MV-141.7 could rapidly diminish CD133+ subpopulations in HCC cells and slow down the tumor growth in vivo. 171 This study also demonstrated the enhanced oncolytic activities of these CD133-specfic viruses when compared with the parental MV-Nse, which is closely related to an oncolytic agent in clinical trials. 171 VB4-845 is an EpCAM-targeted recombinant immunotoxin. 172 Several clinical trials have been conducted to evaluate VB4-845 in patients with urothelial carcinoma, bladder cancer, squamous cell head and neck cancer. 173 In HCC, VB4-845 was found to suppress the sphere-forming ability and reduce the subpopulation of CD133+CD13+ cells. 172 In addition, VB4-845 in combination with 5-FU remarkably suppressed the tumor growth in xenograft models. 172 The administration of anti-CD44 antibody greatly inhibited the activity of CD44 and induced apoptosis of CD90+ MHCC97L and PLC cell lines. 49 A negative correlation between the expression of CD44 and miR-199a-3p was found in primary HCC tissues. 174 The efficiency of miR-199a-3p as a CD44 inhibitor was confirmed in a series of in vitro studies, demonstrating its ability to sensitize the effects of doxorubicin and reduce the invasion of CD44+ HCC cells, suggesting that miR-199a-3p is potent for targeting CD44. 174 CD44 antibody-mediated liposomal nanoparticles have been developed and were shown to specifically target CD44+ HCC cells, leading to improved apoptosis and reduced tumor growth. 175 These nanoparticles also have enabled new approaches to evaluate targeting efficacy and monitor the cancer progression by noninvasive molecular imaging. 175 Ubenimex, an inhibitor of CD13, has been used as an immuno-enhancer in the treatment of multiple myeloma and other solid tumors. 176 In vitro studies have been shown that ubenimex could reverse multidrug resistance (MDR) of HCC cells and improve the sensitivity of established resistant cell lines to cisplatin in a dose-dependent manner. 177 The effects of ubenimex on LCSC regulation remain uncovered and worth further investigation.
Epigenetic alteration
Epigenetic modifications alter the gene expression patterns without changing the primary DNA sequences. 178 Disorder of epigenetic mechanisms, including DNA methylation, histone modification and chromatin remodeling, are involved in the progression of HCC. 179 Multiple observations indicated that LCSC phenotypes were closely associated with epigenetic regulation. Marquardt and colleagues showed that a DNA methyltransferase 1 (DNMT1) inhibitor, zebularine, remarkably increased the tumorigenicity of an isolated side population (SP) fraction from HCC cells. 148 Raggi and colleagues further reported that the adoption of zebularine resulted in enhanced self-renewal and tumorigenic abilities in low-density-grown HCC cells. 180 These zebularine-treated cells also showed high expression levels of CSC- and EMT-related genes, suggesting the crucial role of DNMT1 in LCSCs regulation. 180 SALL4 is a transcriptional repressor through the nucleosome remodeling deacetylase (NuRD) and histone deacetylase (HDAC) complex and is hyperactivated in HCC subtypes with LCSC features. The overexpression of SALL4 displayed increased sphere-forming and invasive capabilities as well as the upregulation of LCSC markers. 181 Clinical investigation demonstrated that SALL4 was positively associated with EpCAM expression and acted as an independent predictor of poor overall survivals in HCC patients. 182 Two HDAC inhibitors, trichostatin A and vorinostat were found to significantly suppress the proliferation of SALL-positive HCC cells. 181 Bmi-1 is a member of the polycomb-group (PcG) family proteins, which function as epigenetic chromatin modifiers in the regulation of self-renewal. 183 Chiba and colleagues reported that Bmi-1 was preferentially expressed in SP cells in Huh7 and PLC/PRL/5 cells and was responsible for the maintenance of tumorigenicity of CD133+ Huh7 cells. 184 Although it remains unknown if Bmi-1 regulates LCSCs through chromatin modulation, the forced silencing of Bmi-1 was reported to reduce the self-renewal and tumorigenicity of SP cells, suggesting that targeting Bmi-1 is a candidate therapeutic strategy for the treatment of LCSCs. 184
MicroRNAs and long noncoding RNAs
miRNA is a major class of nonprotein-coding transcripts which do not encode proteins but may instead function in the posttranscriptional regulation of genes. miRNAs are recognized as key regulators in the self-renewal, differentiation and tumorigenicity of CSCs. Several miRNAs can enhance the LCSC features and act in a promotional fashion in HCC. However, opposite effects could also be found with other miRNAs. For instance, miRNA-449a expression profiles predicted clinical outcomes in HCC patients and was found to correlate with poor prognosis. 185 The exogenous expression of miR-449a increased the CSC features as well as the drug resistance of HCC cells. 185 Mechanically, the overexpression of miR-449a resulted in an enhanced expression of Nanog and a reduced expression level of TCF3, suggesting that miR-449a-TCF3-Nanog axis is a promising target for HCC therapy. 185 Quite the contrary, the overexpression of miR-150 was shown to reduce CD133+ subpopulation in HCC cells and inhibit tumor sphere formation. miR-150 also induced cell cycle arrest and apoptosis in CD133+ HCC cells, accompanies by a remarkably reduction in cyclin D1 and Bcl-2. 186
Long noncoding RNAs (lncRNAs) have been examined in a dozen studies for their roles in the regulation of LCSCs. Wang and colleagues reported that lncTCF7 was required for the maintenance of LCSC self-renewal and tumor propagation via the activation of Wnt signaling. 187 Chen and colleagues showed that the high expression of lncSox4 was associated that HCC events. 188 lncSox4 was required for the self-renewal and tumor initiation of LCSCs via its interactions with STAT3 to initiate the expression of Sox4. 188 Zheng and colleagues demonstrated that lncMEG3 functioned as a tumor suppressor to inhibit cyclin D1 and c-Myc via PKM2 in HCC. 189 This study also indicated that lncMEG3 promoted β-catenin degradation through a PTEN-dependent ubiquitin-proteasome system. 189 Other major findings of miRNAs and lncRNAs in the regulation of LCSCs are listed in Table 3.
The regulatory roles of miRNAs and lncRNAs in LCSCs.
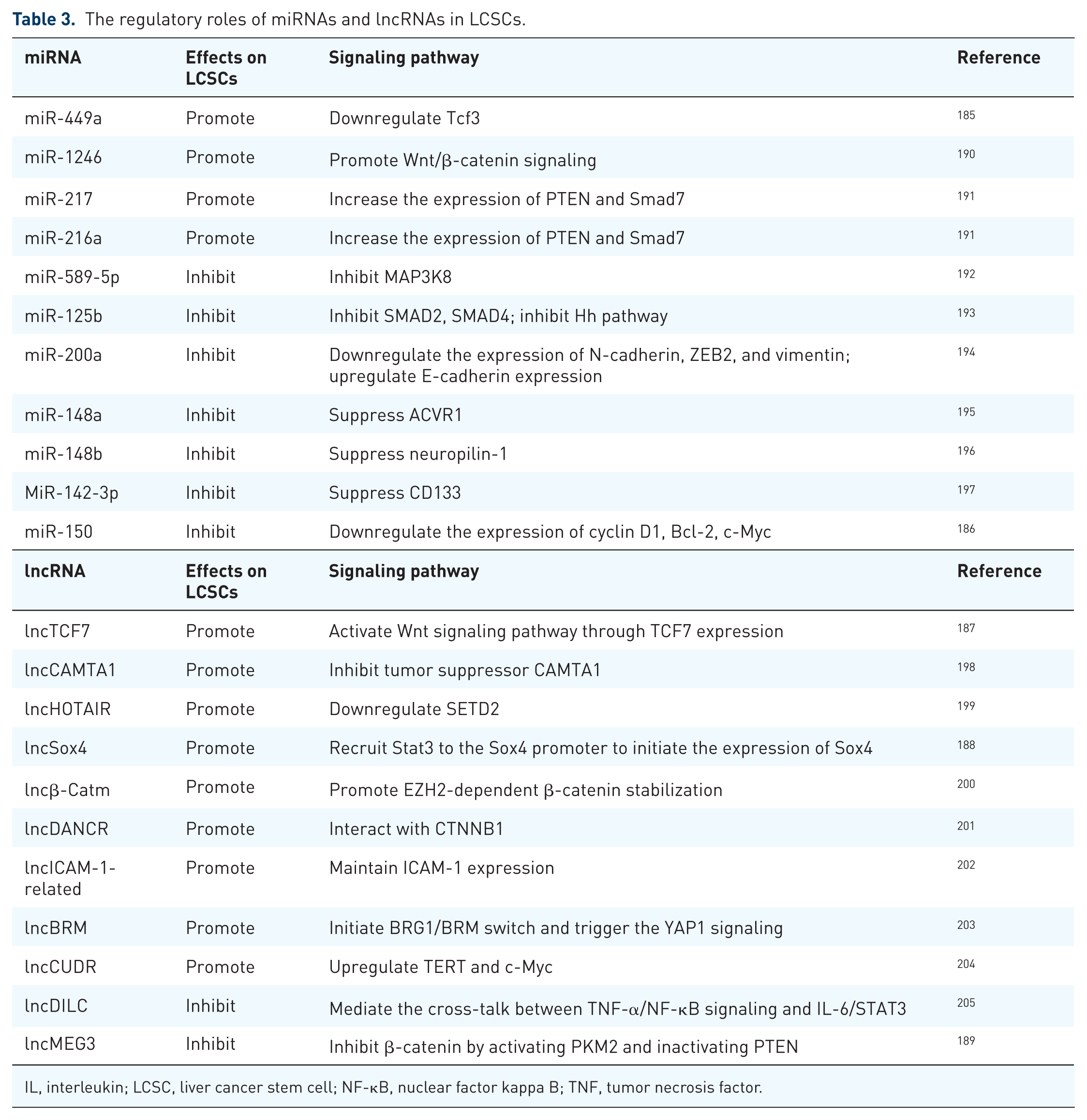
IL, interleukin; LCSC, liver cancer stem cell; NF-κB, nuclear factor kappa B; TNF, tumor necrosis factor.
Targeting autophagy
Autophagy is a self-digestion process in which the removal of endogenous proteins and damaged organelles are achieved by trafficking to lysosome for degradation. 206 Autophagy is viewed as a double-edged sword in hepatocarcinogenesis. Basal autophagy serves as a housekeeping mechanism to maintain cellular homeostasis in normal liver cells; however, unbalanced autophagy turns into the process of tumorigenesis and contributes to the development of HCC under certain microenvironment. 207
Experimental studies proved that autophagy imposes decisions on LCSC induction. In one study, aberrant activation of autophagy was correlated with the transition of Axin2+ cells into Axin2+CD90+ cells which were featured with LCSC characteristics in the development of hepatocarcinoma from liver cirrhosis. 208 The inhibition of autophagy-specific genes or the blockade of autophagy-mediated HGF/Met/JNK or HGF/Met/STAT3 signaling could efficiently decrease the Axin2+CD90+ subpopulation and prevent the hepatocarcinogenetic process. 208 Moreover, Li and colleagues stated that autophagy promoted LCSCs via suppressing p53 expression and retaining it in the cytoplasm. 209 The inhibition of autophagy with 3-methyladenine and bafilomycin A1 significantly reduced the CD133+ proportion in HCC cells and impaired the sphere-forming abilities, suggesting that the use of autophagy inhibitors is an effective approach against LCSCs. 209
Targeting the ABC transporter
Numerous studies have demonstrated that CSCs exhibit enhanced expression of ABC transporters when compared with nonstem populations. 19 ABC transporters have been considered to be involved in the regulation of cancer stem cell physiology and drug resistance. A member of the ABC transporter, ABCG2, is well known as a tentative LCSC marker in HCC, which highlighted its potential role in the maintenance of cancer stemness. As a multidrug-resistance transporter, ABCG2 has been shown to elevate the CSC-related malignant characteristics and chemoresistance. 210 Consistently, ABCG2 was found to be abundantly expressed in enriched CD90+CD133+ LCSCs and positively associated with the acquisition of drug resistance. 70 Several ABCG2 inhibitors for clinical usage have been developed as therapeutics agents in various cancer types. In HCC, the phase II study of an ABCG2 inactivator, gefitinib, in advanced unresectable HCC was completed in 2013 (ClinicalTrials.gov Identifier: NCT00071994). However, this single agent failed to exert potent activities in advanced HCC. Gefitinib was administered at 250 mg daily in a two-stage designed study. With a median follow up of 13.2 months, the median overall survival and progression-free survival of the enrolled 31 patients was 6.5 months [95% confidence interval (CI): 4.4, 8.9] and 2.8 months (95% CI: 1.5, 3.9) respectively in the first stage. No patient had a complete response. Only one patient experienced a partial response and seven patients displayed stable disease. Based on these results and the reason that the criteria for second-stage accrual was not met, the investigators have thus concluded that gefitinib is not active in advanced HCC. 211
The prevalent expression and distinguished functions of ABC transporters in CSCs also addressed its implication on LCSCs identification. The SP was described as a small subpopulation of cells with CSC-like properties and high ABC transporters expression profiles. These cells are isolated due to their improved ability to efflux the Hoechst dye via the ABCG2 membrane transporters than the remaining HCC cells termed as non-SP populations. 212 The SP population cells exhibit strong CSC properties including spheroid-forming, proliferation, invasion, migration, tumorigenesis and the overexpression of stem cells markers including Klf4, Sox2, Sox9 and SALL4 are evidenced in SP cells.213,214 SP cells in HCC appear to be linked to some miRs. For example, miR-200a was downregulated in SP cells, contributing to metastasis in HCC, and the overexpression of miR-200a decreased the levels of metastasis-related markers as well as the expression of ZEB2 in SP cells. 215 In contrast with miR-200a, C19MC miRNAs (the miRNA cluster on chromosome 19) was upregulated in SP cells and associated with poorer prognosis and advanced disease in HCC. 216 The enhanced invasion and migration of SP cells may be attributed to the overexpression of cyclooxygenase-2 (COX-2), as the administration of COX-2 inhibitor celecoxib can lead to significantly reduce the migration and invasion of SP cells in a dose-dependent manner, along with the upregulation of antimetastasis-related proteins PDCD4 and PTEN. 217 Therefore, targeting key molecules in SP cells can be a promising strategy to eradicate LCSCs.
Discussion and conclusion
There is growing evidence that tumor initiation may be driven by a subset of tumor cells which have the capacities of self-renewal, differentiation and resistance to traditional therapies. The LCSC nature of tumorigenesis provoked deeper thinking and investigations for its roles in HCC. The study of LCSCs rely on the identification and isolation of subpopulations by putative CSC surface markers. Mounting data have suggested that the existence of LCSCs manipulates the tumor initiation, metastasis, therapeutic resistance as well as HCC recurrence and lays a foundation for the development of LCSC-targeted therapies.
The recent advances of LCSC biology have accelerated the study aimed at CSC eradiation. Several therapeutic strategies are being developed to target CSCs. Experimental studies have proved that targeting core signaling cascades in CSCs has satisfactory results. The Wnt, Notch, Hh, and TGF-β pathway inhibitors suppress LCSC self-renewal, metastasis and tumorigenicity in vitro and in vivo. Successful preclinical investigations led to the development of clinical studies with these inhibitors. Among those, Wnt inhibitor OMP-54F28, Hh inhibitor LED225 and TGF-β signaling inhibitor galunisertib have been enrolled in clinical trials alone or in combination with other therapeutic agents for the treatment of HCC. 106 Given the cross-talk between cancer cells and CSCs, the intricate CSC microenvironment could not only supply tumor growth signals but also take part in the acquisition of therapeutic resistance. Treatments designed to target CSC microenvironment, singly or in combination with other drugs, represent an efficient strategy for the treatment of LCSCs. Targeting LCSC surface markers provides us a straightforward perspective to address the CSC population. LCSC marker-targeted therapeutic agents have been examined in series of studies and produced positive results. Epigenetic control of gene expression is involved in every step of tumorigenesis, propagation and survival. Moreover, the heritable and reversible features of epigenetic alteration make it a critical biological function. Epigenetic therapies were found to significantly reduce the CSC characteristics. However, epigenetic modulations in cancer seldom target specific genes, which could result in nonspecific alteration of gene actions. And current studies lack evidence to evaluate their safety for the treatment of LCSCs. Given the pivotal role of miRNAs and lncRNAs in the regulation of CSC phenotype and HCC progression, the development of miRNA/lncRNA-targeted therapies will be likely to achieve great therapeutic effects.
In this review, we have summarized the functions of LCSCs and the recent advances in the recognition of anti-LCSCs agents (Figure 1). Targeting of LCSCs via the Wnt, Notch, Hh, and TGF-β pathways as well as targeting the CSC niche, surface markers, autophagy process and ABC transporters are promising strategies to eliminate LCSCs and important additions to traditional anti-HCC therapies. Although, current clinical results have not yet elucidated to bolster the findings from preclinical studies, extensive investigations of these treatment regimens provide new insights into the treatment of HCC. The development of LCSC-targeted agents will shift the paradigm of HCC treatment and might be a breakthrough in the fight against metastasis, recurrence and therapeutic resistance. In the meantime, targeting the CSC compartment should have limited toxicity to normal stem cells. It should be noted that the normal stem cell niche in adult somatic tissues plays an essential role in maintaining the stem-like state and regulating cell fate. 218 Due to the similarities between CSCs and normal stem cells, drugs that target the CSC niche may also give rise to severe health issues. With an emerging understanding of LCSC biology, it is anticipated that improved targeted LCSC therapies will be developed in the future.

The functions and potential therapeutic strategies of LCSCs.
Footnotes
Acknowledgements
We thank Ms Christina Lou for her editing support.
Funding
This study was supported by grants from the Research Grants Council of the Hong Kong Special Administrative Region (No. 14109516 and 14117015), the National Natural Science Foundation of China (No.81472339) and a CUHK Direct Grant (2015.1.083).
Conflict of interest statement
The authors declare that there is no conflict of interest.