
Abstract
The cyclin D/cyclin-dependent kinases 4 and 6 (CDK4/6)–retinoblastoma protein (RB) pathway plays a key role in the proliferation of both normal breast epithelium and breast cancer cells. A strong rationale for inhibiting CDK4/6 in breast cancers has been present for many years. However, potent and selective CDK4/6 inhibitors have only recently become available. These agents prevent phosphorylation of the RB tumor suppressor, thereby invoking cancer cell cycle arrest in G1. CDK4/6 inhibitors have transited rapidly from preclinical studies to the clinical arena, and three have already been approved for the treatment of advanced, estrogen receptor (ER)-positive breast cancer patients on account of striking clinical trial results demonstrating substantial improvements in progression-free survival. ER-positive breast cancers harbor several molecular features that would predict their sensitivity to CDK4/6 inhibitors. As physicians gain experience with using these agents in the clinic, new questions arise: are CDK4/6 inhibitors likely to be useful for patients with other subtypes of breast cancer? Are there other agents that could be effectively combined with CDK4/6 inhibitors, beyond endocrine therapy? Is there a rationale for combining CDK4/6 inhibitors with novel immune-based therapies? In this review, we describe not only the clinical data available to date, but also the biology of the CDK4/6 pathway and discuss answers to these questions. In particular, we highlight that CDK4 and CDK6 govern much more than the cancer cell cycle, and that their optimal use in the clinic depends on a deeper understanding of the less well characterized effects of these enzymes.
Keywords
Introduction
Dysregulated cellular proliferation is a characteristic of all human cancers, and the propensity for tumor cells to sustain aberrant proliferative signaling has been heralded as a ‘hallmark of cancer’. 1 In normal tissues, cell proliferation is tightly regulated by the cell cycle machinery, a group of proteins that control a cell’s orderly procession from one phase of the cell cycle to the next. In breast cancer, much attention has been given to particular members of the cell cycle machinery – the D-type cyclins and their partner kinases, cyclin-dependent kinase 4 (CDK4) and CDK6. Indeed, a wealth of preclinical research has shown that tumor cell proliferation in many breast cancers is underpinned by hyperactivity of the cyclin D–CDK4/6 axis,2–4 making pharmacological blockade of this axis an attractive therapeutic strategy.
Potent, selective, orally bioavailable inhibitors of CDK4/6 have only become available as cancer therapeutics in the last decade. By directly blocking the activity of the cyclin D–CDK4/6 holoenzyme, these agents act to restrain proliferation of sensitive tumor cells, in particular preventing cell cycle progression from the G1 to the S phase of the cell cycle (see below and Figure 1). In sensitive cells, CDK4/6 inhibition typically induces a phenotype resembling cellular senescence, 5 consistent with the critical role of the retinoblastoma (RB) tumor suppressor in mediating senescence. 6

The role of the cyclin D1–CDK4/6–RB pathway in breast cancer.
In human breast cancer, the subtype for which CDK4/6 inhibition has the strongest rationale is estrogen receptor (ER)-positive disease. These cancers almost always retain RB function at presentation, meaning that the principal pathway upon which these agents act is intact. 7 Moreover, CCND1 (encoding cyclin D1) is a direct target gene of the ER, and is thus often expressed at high levels in ER-positive cancers. Preclinically, strong synergy has been observed when CDK4/6 inhibitors are added to standard anti-estrogen therapies, 7 and large, randomized clinical trials have confirmed that the addition of CDK4/6 inhibitors to hormonal therapy is a valuable clinical approach.8–11 Three CDK4/6 inhibitors have now been approved by the FDA for the treatment of ER-positive metastatic breast cancer: palbociclib (PD0332991), ribociclib (LEE011) and abemaciclib (LY835219). The addition of these agents to endocrine therapy has resulted in the longest improvement in progression-free survival (PFS) seen to date in this subtype of breast cancer.8–11
We are only just beginning to fully understand how CDK4/6 inhibitors work, and as more preclinical and clinical studies are published, new questions arise. Are there other agents that we could be combining with CDK4/6 inhibitors for the treatment of breast cancer? Which patients are most likely to benefit from these drugs? Are all approved CDK4/6 inhibitors the same or are there intrinsic differences in their mechanisms of activity? Is there a role for the use of these drugs in other subtypes of breast cancer? One particularly difficult challenge has been to use our growing knowledge of CDK4/6 pathway biology to elucidate predictors of drug response and resistance in patients. The purpose of this review is to summarize the background and latest evidence for the use of CDK4/6 inhibitors in breast cancer, and discuss some of the unanswered questions about the use of these agents in clinical practice.
The cyclin D–CDK4/6-retinoblastoma pathway
In normal mammary tissue, cyclin D1 and CDK4 in particular are important for luminal epithelial proliferation, and these dependencies are often upheld in luminal breast cancers.3,4,12,13 Cyclin D1 binds to CDK4, and the protein complex is stabilized by proteins such as p21, rendering it an active holoenzyme. 14 The holoenzyme then monophosphorylates the RB protein. Phosphorylation of RB depresses the E2F family of transcription factors, enabling the expression of cyclin E, another S phase cyclin. Cyclin E then binds to and activates CDK2, which hypherphosphorylates RB, further liberating E2F transcription factors and facilitating the expression of a wide variety of genes that promote transit from G1 to S phase. 15 Small molecule CDK4/6 inhibitors act primarily by blocking RB phosphorylation and thus inducing G1 cell cycle arrest and a phenotype resembling cellular senescence. 16 In vitro, tumor cells with a non-functional RB pathway are resistant to CDK4/6 inhibitors,7,17 presumably because they lack the canonical target of these agents.
The enzymatic activity of CDK4/6 is regulated by several mechanisms. 18 First, several mitogenic signaling pathways that are active in breast cancers positively regulate CDK4/6 activity by increasing CCND1 expression and/or increasing cyclin D1 protein stability.19–21 These include signaling through receptor tyrosine kinases (such as EGFR and HER2), the PI3K–AKT–mTOR axis and the ER (Figure 1).19–21 With respect to receptor tyrosine kinase signaling, mouse models have shown that cyclin D1 is an absolute requirement for the formation of mammary adenocarcinomas driven by ERBB2, and that cyclin D1/CDK4 also plays an important role in the growth of established ERBB2-driven tumors.3,16,20 Furthermore, cyclin D1 is a direct transcriptional target of ER, and estrogens promote the transit of ER-positive breast cancer cells from G1 to S phase. 22 Conversely, anti-estrogen therapies such as tamoxifen, aromatase inhibitors, and fulvestrant reduce cyclin D1 expression and hence induce G1 cell cycle arrest. 23 Notably, cyclin D1 can also activate expression of ER target genes in an estrogen-independent manner. 24 Finally, approximately 15% of breast cancers demonstrate amplification of CCND1 itself, and these tumors tend to have higher levels of cyclin D1 protein as well.25,26 This serves as yet another potential way to stimulate CDK4 activity in breast cancers.
In addition to this multitude of mechanisms by which CDK4 activity can be stimulated in breast cancers, cells also harbor a number of endogenous proteins that directly inhibit CDK4/6 activity. The most important of these endogenous CDK4/6 inhibitors are the INK4 proteins (p16INK4a, p15INK4b, p18INK4c, and p19INK4d). These proteins bind specifically to CDK4 and CDK6 and inhibit their catalytic subunits.14,27 A small proportion of breast cancers demonstrate deep deletion of CDKN2A, the gene encoding p16. Theoretically, such tumors would be expected to have higher baseline CDK4/6 activity and thus potentially be more sensitive to CDK4/6 inhibitors. As is discussed below, however, this remains a question of controversy. 8
CDK4/6 inhibitors in metastatic ER-positive breast cancer
CDK4/6 inhibitors have shown both preclinical and clinical activity in ER-positive breast cancer, and data suggest synergy when combining anti-estrogen therapy with CDK4/6 inhibitors.7,28 Significant improvements in PFS seen in clinical trials (Tables 1 and 2) have led to their approval in metastatic ER-positive, HER2-negative breast cancer patients in combination with endocrine therapy, or in the case of abemaciclib also as a single agent. It is worth noting that the three approved CDK4/6 inhibitors present distinct relative potencies for CDK4 and CDK6 inhibition, pharmacokinetics, dosing schedules and toxicity profiles.
Randomized phase II/III clinical trials of CDK4/6 inhibitors as first-line treatment of advanced ER-positive breast cancer.

In patients with measurable disease at baseline.
CBR, clinical benefit rate; NR, not reached; NSAI, nonsteroidal aromatase inhibitor; ORR, objective response rate; PFS, progression-free survival.
Major clinical trials of CDK4/6 inhibitors in patients with advanced ER-positive breast cancer who had previously progressed on endocrine therapy.

Not yet reported.
CBR, clinical benefit rate; CT, chemotherapy; ET, endocrine therapy; MBC, metastatic breast cancer; ORR, objective response rate; PFS, progression-free survival.
In clinical practice, palbociclib is typically administered at a starting dose of 125 mg daily 3 weeks on, 1 week off, in conjunction with endocrine therapy. This dose was initially tested in patients with RB-expressing (as determined by IHC) metastatic breast cancer using palbociclib as a single agent in a phase II, single-arm study. 32 Patients in this study were heavily pretreated and the overall activity observed was modest [objective response rate (ORR) 5%, clinical benefit rate (CBR) 19%, and median PFS 3.7 months]. 32 However, in the subsequent randomized phase II PALOMA-1/TRIO-18 trial, the combination of letrozole plus palbociclib yielded an impressive 10-month improvement in PFS (20.2 versus 10.2 months, HR, 0.49, p = 0.0004) in patients with advanced ER-positive, HER2-negative breast cancer, when given as first-line therapy. 8 These results led to the phase III PALOMA-2 trial which confirmed this significant and remarkable benefit in PFS (see Table 1). 11 As described in Table 2, palbociclib also shows efficacy in endocrine therapy-resistant metastatic breast cancer, improving PFS when added to fulvestrant as second-line therapy. 33
Based on the findings of the phase III MONALEESA-2 trial 9 (Table 1), the FDA approved ribociclib to be used in combination with an aromatase inhibitor as initial therapy for the treatment of postmenopausal women with ER-positive, HER2-negative advanced breast cancer. Recently, the results of the phase III MONALEESA-7 trial were presented, specifically addressing the question of CDK4/6 efficacy in pre- or perimenopausal women (Table 1). 35 All women in the study received ovarian function suppression together with oral endocrine therapy (tamoxifen or an aromatase inhibitor) ± ribociclib, and results were very similar to those observed in the postmenopausal trials.8–11 Of note, this was the first major study to include tamoxifen as one of the endocrine therapy partners to CDK4/6 inhibition, and a similar improvement in PFS was observed with either endocrine regimen.
Like the other CDK4/6 inhibitors, randomized phase III trials have demonstrated the benefit of adding abemaciclib to hormonal therapy. In patients with advanced ER-positive breast cancer, both ORR and PFS were significantly improved, not only for initial therapy in postmenopausal women (MONARCH 3) (Table 1), 10 but also in combination with fulvestrant in patients previously treated with endocrine therapy (MONARCH 2) (Table 2). 41 Strikingly, however, abemaciclib also demonstrates significant activity as a single agent. In the phase II single-arm MONARCH 1 trial (Table 2), 42 abemaciclib monotherapy yielded an ORR of 19.7% and CBR of 42.4% in a heavily pretreated population with ER-positive, HER2-negative metastatic breast cancer. Notably, the median time to response was 3.7 months and the median PFS and overall survival (OS) were 6 and 17.7 months, respectively. Given that CDK4/6 inhibitors act primarily by inducing cell cycle arrest, mechanisms behind this response are unclear. However, preclinical studies suggest that a portion of breast cancers might undergo apoptosis in response to abemaciclib 17 and/or that abemaciclib might induce an anti-tumor immune response that underpins some of its activity and explains tumor regression.43,44
Table 1 summarizes the phase II/III randomized clinical trials of CDK 4/6 inhibitors as first-line treatment for advanced ER-positive, HER2-negative breast cancer. Results are consistent across all these randomized trials in terms of efficacy, with similar median PFS, hazard ratios and patient populations. Sixty percent of patients had visceral disease at baseline and about 40% had metastatic de novo breast cancer. Remarkably, these trials demonstrate ORRs of over 50%, similar to that achieved with first-line chemotherapy. Table 2 summarizes major trials of CDK4/6 inhibition in patients with advanced HR-positive breast cancer who have relapsed or progressed during endocrine therapy. Again, hazard ratios across trials are similar. There are, however, some differences with respect to patient populations in these trials, which might in part explain the differences in absolute PFS. In MONARCH-2, 41 prior chemotherapy was not permitted and just one prior endocrine therapy was allowed, whereas in PALOMA-3, 33 approximately one-third of patients had received prior chemotherapy and any number of prior endocrine therapies was permitted.
The remarkable results observed with CDK4/6 inhibitors in these trials have led to the initiation of many other studies. 45 The PEARL trial (ClinialTrials.gov identifier: NCT02028507) is a randomized phase III study that compares palbociclib plus endocrine therapy versus capecitabine chemotherapy in postmenopausal patients with metastatic ER-positive, HER2-negative breast cancer resistant to previous nonsteroidal aromatase inhibitor therapy. In addition, numerous clinical trials are also evaluating the efficacy of CDK4/6 inhibitors in settings beyond advanced disease and in subtypes other than ER-positive disease, as is described below.
CDK4/6 inhibition in early-stage ER-positive breast cancer
The exciting results observed with CDK4/6 inhibitors in the treatment of advanced ER-positive, HER2-negative breast cancer have triggered the evaluation of these agents in the early-stage setting. Indeed, there is an urgent need to improve the efficacy of adjuvant endocrine therapy, given that a considerable number of patients with ER-positive breast cancer experience disease recurrence, and that the risk of recurrence remains even decades after initial diagnosis. 46 First, each of the three agents is now being tested in a randomized adjuvant phase III study for patients with high-risk, ER-positive disease. These are summarized in Table 3. Each study has slightly different inclusion criteria, but all are comparing the efficacy of a CDK4/6 inhibitor as an adjunct to standard endocrine therapy.
Randomized phase III clinical trials evaluating CDK 4/6 inhibitors in early-stage ER-positive/HER2-negative breast cancer (ClinicalTrials.gov January 2018).
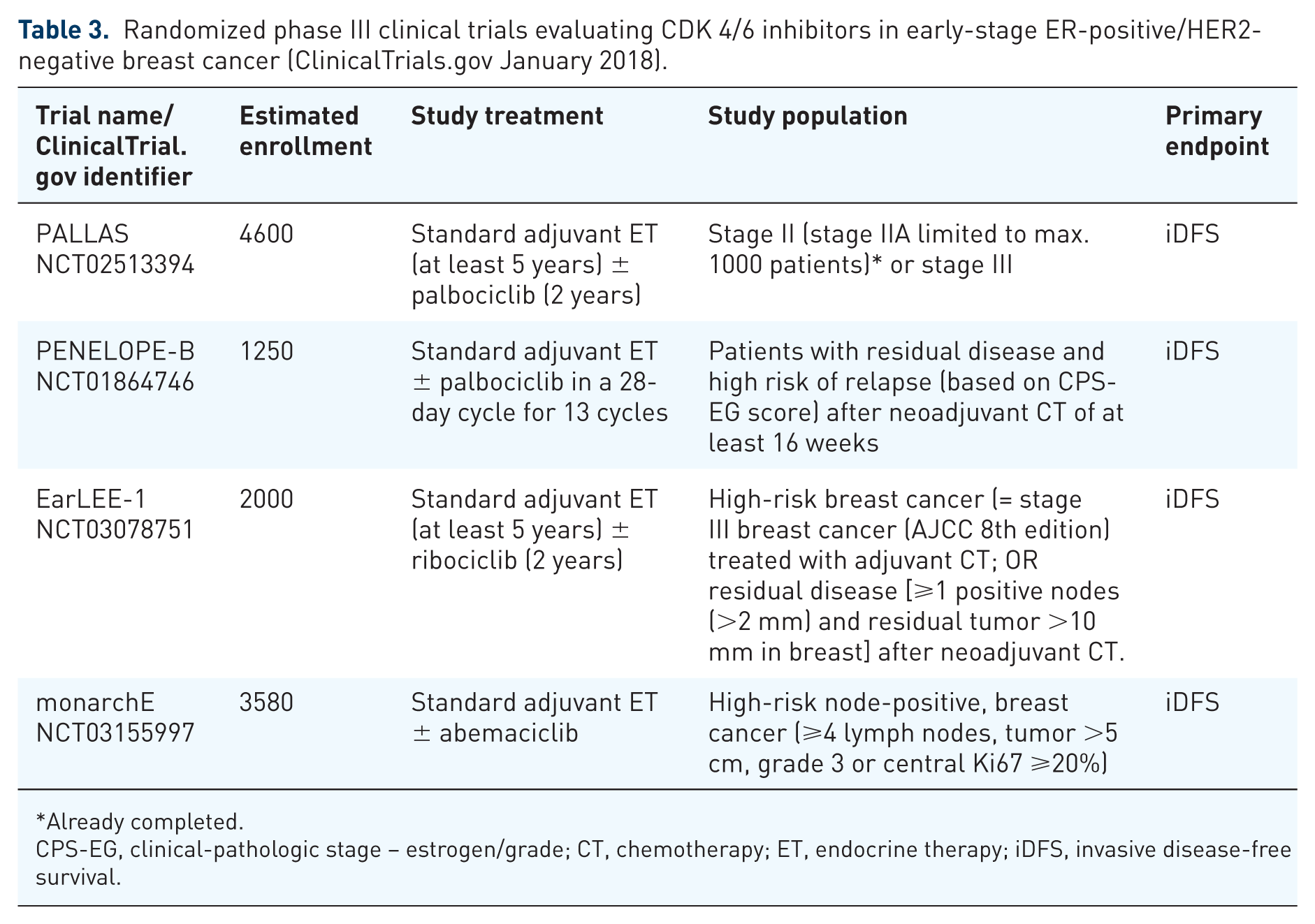
Already completed.
CPS-EG, clinical-pathologic stage – estrogen/grade; CT, chemotherapy; ET, endocrine therapy; iDFS, invasive disease-free survival.
Several neoadjuvant trials have also been planned, and some have already been reported. The trials have typically been designed to answer translational science questions, taking advantage of the ability to perform serial biopsies of primary tumors during treatment. The single-arm phase II NeoPalAna trial evaluated the anti-proliferative effect of palbociclib. 47 Fifty patients with stage II–III ER-positive, HER2-negative breast cancer were enrolled. Treatment consisted of anastrozole for 4 weeks (with goserelin if premenopausal), followed by the addition of palbociclib for four 28-day cycles. Serial biopsies were performed and analyzed for Ki67, gene expression and mutation profiles. The primary endpoint was complete cell cycle arrest (CCCA), defined as Ki67 ⩽2.7%, as assessed 2 weeks after initiation of palbociclib. Palbociclib enhanced cell cycle control over anastrozole monotherapy, and this effect was observed across various subgroups, including grade 3 tumors, those with PIK3CA mutations and tumors in pre- and postmenopausal women. Interestingly, palbociclib’s anti-proliferative effect was rapidly lost when CDK4/6 treatment was held prior to surgery, suggesting that the “senescence” induced by these agents is not truly irreversible, but rather depends on ongoing drug treatment. In the phase II neoMONARCH trial, 220 postmenopausal women with stage I–IIIB ER-positive, HER2-negative breast cancer were randomized, after a baseline biopsy, to anastrozole, abemaciclib or the combination for 2 weeks, after which another core biopsy was performed. 48 Patients then continued on abemaciclib and anastrozole for a further 14–22 weeks. In this trial, abemaciclib alone or in combination with anastrozole significantly reduced Ki67 expression compared to anastrozole alone after 2 weeks of treatment (primary endpoint based on geometric mean change in Ki67 and CCCA defined as Ki67 ⩽2.7%), and induced a profound cell cycle arrest and changed gene expression in a manner suggestive of cellular senescence. Abemaciclib treatment also appeared to induce histologic changes suggestive of increased tumor differentiation and increased lymphocyte infiltration in some cases. 48
The combination of ribociclib plus letrozole in ER-positive, HER2-negative breast cancer is being assessed in several phase II neoadjuvant trials. In the FELINE study (ClinicalTrials.gov identifier: NCT02712723), patients are randomized to either placebo plus letrozole or ribociclib plus letrozole (ribociclib at two different dosing schedules). In the CORALLEEN study (ClinicalTrials.gov identifier: NCT03248427), luminal B (as assessed by PAM50 intrinsic subtyping) HER2-negative breast cancer patients are randomized to either neoadjuvant multi-agent chemotherapy or to ribociclib plus letrozole for 6 months. In another phase II trial, luminal breast cancer patients are randomized to either weekly paclitaxel or endocrine treatment in combination with palbociclib for 12 weeks; after that, treatment is switched in a crossover design (ClinicalTrials.gov identifier: NCT02603679). The PELOPS trial (ClinicalTrials.gov identifier: NCT02764541) is an open-label phase II neoadjuvant trial evaluating the combination of palbociclib and endocrine therapy within cohorts of HR-positive breast cancer patients with invasive lobular and ductal carcinoma. Finally, neoadjuvant treatment with CDK4/6 inhibitors has also been tested in ER-positive, HER2-positive disease, as described below. 49
CDK4/6 inhibitors for other breast cancer subtypes
HER2-positive breast cancer
Despite the successes of anti-HER2 targeted agents for HER2-positive breast cancers, drug resistance remains a significant challenge for a subset of patients that requires novel therapeutic approaches. 20 As described above, cyclin D1 expression lies directly downstream of HER2 signaling and the initial preclinical trials demonstrating the critical importance of cyclin D1–CDK4 in breast tumorigenesis were performed in ERBB2-driven tumors.3,16 As such, there is a strong rationale to test CDK4/6 inhibitors in HER2-positive breast cancers. In support of this rationale, early preclinical studies showed that several HER2-positive breast cancer cell lines are markedly sensitive to CDK4/6 inhibition in vitro. 7 Moreover, several groups have demonstrated synergy between anti-HER2 therapy and CDK4/6 inhibitors7,50 and that CDK4/6 inhibition can specifically overcome acquired resistance to anti-HER2 therapy. 20 In preclinical experiments, tumor cells surviving HER2-blockade retain high expression of cyclin D1, and targeting these cells with CDK4/6 inhibitors resensitizes them to anti-HER2 therapy not only by reducing RB phosphorylation but also suppressing mTORC1/S6K/S6RP activity and increasing tumor cell dependence on EGFR family kinases. 20 Indeed, combined HER2 and CDK4/6 inhibition has a synergistic effect on suppressing tumor cell proliferation through enhancement of G1 arrest, both in vitro and in vivo. Finally, CDK4/6 inhibition can delay recurrence of HER2-driven breast cancers in mouse models. 20
The early clinical data also support the notion of using CDK4/6 inhibitors in HER2-driven breast cancers. In the initial phase II trial evaluating the efficacy of palbociclib as a single agent, 32 two patients had ER-positive, HER2-positive cancer. Of those, one patient experienced a partial response (PR) and the other had stable disease (SD) lasting 5 months, without concurrent HER2-directed therapy. Likewise, all 11 ER-positive, HER2-positive breast cancer patients included in a phase I study of abemaciclib experienced disease control, including a 36% rate of PR. 39 In contrast, there were no responses observed among patients with ER-negative, HER2-positive tumors.
In the neoadjuvant setting, the phase II open-label NA-PHER trial explored the activity of a four-drug regimen comprising trastuzumab, pertuzumab, palbociclib and fulvestrant in HER2-positive, ER-positive breast cancer patients and reported encouraging antitumor activity. 49 This chemotherapy-free regimen induced a significant reduction in Ki67 expression (defined as the percentage of positively staining cells within the invasive margin in the examined area) at week 2 and at surgery, after 16 weeks of treatment. At baseline, the geometric mean Ki67 expression was 31.9 (SD 15.7) versus 4.3 (15.0; paired test p < 0.0001) at week 2 and 12.1 (20.0; p = 0.013) at the time of surgery. Remarkably, 50% of patients achieved a complete clinical response and 27% achieved pathological complete response (pCR) in breast and lymph nodes. 49 This pCR rate compares favorably to that observed in similar patients treated with chemotherapy and anti-HER2 therapy, and speaks to the crosstalk between CDK4/6 and HER2 signaling in breast cancer cells. 51 Table 4 outlines the ongoing randomized trials evaluating CDK4/6 inhibitors in advanced HER2-positive breast cancer patients. Included are two global, randomized trials – the MonarcHER study (ClinicalTrials.gov identifier: NCT02675231) evaluates the role of abemaciclib in patients with pretreated metastatic disease, and the PATINA study (ClinicalTrials.gov identifier: NCT02947685) explores the benefits of adding palbociclib to standard first-line metastatic therapy. In addition, data from stage 1 of the PATRICIA (SOLTI 13-03) trial were recently reported. 52 Thus far, this phase II trial has demonstrated that in pretreated patients with HER2-positive, ER-positive advanced breast cancer, objective responses are observed with palbociclib and trastuzumab. 52 Other non-randomized phase Ib/II studies in advanced HER2-positive breast cancer include those combining palbociclib and T-DM1 (ClinicalTrials.gov identifier: NCT01976169), palbociclib, trastuzumab, pertuzumab and anastrozole (ClinicalTrials.gov identifier: NCT03304080), ribociclib with trastuzumab or T-DM1 (ClinicalTrials.gov identifier: NCT02657343), and palbociclib with tucatinib and letrozole (ClinicalTrials.gov identifier: NCT03054363).
Ongoing randomized phase II/III clinical trials evaluating CDK4/6 inhibitors in advanced HER2-positive breast cancer (ClinicalTrials.govJanuary 2018).

BC, breast cancer; CPC, chemotherapy of physician’s choice; CT, chemotherapy; PFS, progression-free survival.
Aside from awaiting the results of definitive trials, the pressing questions pertaining to CDK4/6 inhibitor use in HER2-positive breast cancer relate to defining subtypes that might benefit most. The large randomized trials are restricted to patients with ER-positive, HER2-positive tumors, in part because these showed the greater response rates in early studies, 39 and in part because these are more likely to demonstrate luminal biology and perhaps be more cyclin D1–CDK4 dependent. However, given the direct connection between cyclin D1 and HER2, there is a biologic rationale to consider CDK4/6 inhibitors in patients with ER-negative, HER2-positive disease (at least in those tumors that retain RB function). The PATRICIA trial 52 is including some such patients, and results will be very informative. Finally, once trials are completed, analysis should also be performed comparing results across patients with tumors of different PAM50 subtypes. Again, the PATRICIA trial has provided initial suggestions that certain subtypes might demonstrate better prognosis on CDK4/6–HER2 combinations; particularly that the luminal subtype predicts a better PFS compared to non-luminal (10.37 versus 3.53 months, HR 0.34, 95% CI; 0.13–0.92; p = 0.033). However, these data do not distinguish between disease behavior per se and response to CDK4/6 inhibitors specifically. 52
Triple-negative breast cancer (TNBC)
TNBC is a molecularly heterogeneous disease in urgent need of effective targeted therapies. Traditionally, TNBC has been considered a poor candidate for CDK4/6 inhibitor therapy, as tumors often demonstrate loss of expression of the RB protein (either due to genomic loss or through other mechanisms), 53 or high expression of cyclin E – both of which would be expected to confer resistance to treatment with CDK4/6 inhibitors. Moreover, many TNBC cell lines are insensitive to CDK4/6 inhibition in vitro. 7 However, recent studies have found that some TNBCs might be sensitive to CDK4/6 inhibition. In fact, Asghar and colleagues 54 demonstrated that the luminal androgen receptor (LAR) subtype of TNBC was highly sensitive to CDK4/6 inhibition both in vitro and in vivo. In contrast, basal-like and mesenchymal TNBC cell lines were resistant to CDK4/6 inhibition. They also identified temporal dysregulation of cyclin E expression (and hence increased CDK2 activity) as a possible mechanism by which RB wildtype TNBCs might escape the effects of CDK4/6 inhibitors. 54 In addition, dual blockade of CDK4/6 and PI3K has demonstrated substantial activity not only in LAR and mesenchymal-stem (MSL) subgroups, 54 which frequently harbor mutations in the PI3K catalytic subunit of the PIK3CA gene, 55 but also in a variety of RB1-wildtype TNBC models, regardless of PIK3CA mutation status. 56
Another intriguing observation demonstrated in preclinical TNBC models is that inhibition of CDK4/6 can block breast cancer metastasis. 57 Liu and colleagues identified the deubiquitinase DUB3 as a new target of CDK4/6. CDK4/6-mediated activation of DUB3 is essential to deubiquitinate and stabilize SNAIL1, a transcription factor that promotes epithelial–mesenchymal transition (EMT) and, therefore, invasiveness. Collectively, these results provide rationale for testing CDK4/6 inhibitors for some TNBCs, and for identification of markers of sensitivity. Indeed, there are ongoing clinical trials evaluating abemaciclib as a single agent in metastatic RB-positive TNBC (ClinicalTrials.gov identifier: NCT03130439) or the combination of palbociclib or ribociclib with bicalutamide in AR+ TNBC (ClinicalTrials.gov identifiers: NCT02605486 and NCT03090165, respectively).
Novel CDK4/6 inhibitor combinations
Combinations with PI3K/AKT/mTOR inhibitors
There is profound crosstalk between the CDK4/6 and the PI3K–AKT–mTOR pathways, including many negative feedback loops, providing strong rationale for combining inhibitors through both axes to inhibit tumor growth (Figure 1).19–21,58 Moreover, it has been shown that early adaptive resistance to CDK4/6 inhibitors in ER-positive breast cancer cells might depend on a compensatory PI3K-dependent activation of non-canonical cyclin D1–CDK2, and hence recovery of RB phosphorylation. 59 Combined treatment with PI3K and CDK4/6 inhibitors has been shown to overcome single-agent CDK4/6 inhibitor resistance in ER-positive breast cancer cells, due to downregulation of cyclin D1, inducing not only a profound cell cycle arrest but also apoptosis.21,59 Moreover, the triplet combination of fulvestrant and dual inhibition of CDK4/6 and PI3K was more effective than either doublet both in vitro and in vivo. 59
A synergistic interaction has been also observed between PI3K and CDK4/6 inhibitors in TNBC. Both the combination of palbociclib with taselisib (in PIK3CA-mutant TNBC cells) or of ribociclib with alpelisib (in a variety of TNBC preclinical models) demonstrated greater efficacy, in terms of cell cycle arrest and apoptosis, than either drug alone.54,56 Inhibition of PI3K signaling sensitized to CDK4/6 inhibition, in part by suppressing post-mitotic CDK2 activity and therefore inducing a quiescent state in which CDK4/6 activity is required to initiate the cell cycle. 54
Based on such data, trials examining CDK4/6–PI3K and CDK4/6–mTOR inhibitor combinations are underway. Some phase I/II clinical trials in advanced HER2-negative breast cancer include the combination of ribociclib, fulvestrant and BKM 120 or BYL719 (ClinicalTrials.gov identifier: NCT02088684). Other trials are exploring the combination of endocrine therapy with CDK4/6 and mTOR inhibitors in patients who have progressed on a CDK4/6 inhibitor (see section: Continuing CDK4/6 inhibition beyond progression). Data from these trials are awaited, and the first hurdle will be to establish that such regimens can be tolerated without prohibitive toxicity.
Combinations with immune checkpoint inhibitors
Recently, a number of preclinical studies have been published suggesting that CDK4/6 inhibitors not only induce tumor cell cycle arrest, but also incite an anti-tumor immune response. The mechanisms behind this include enhanced tumor cell antigen presentation due to heightened expression of endogenous retroviral sequences resulting in tumor cell interferon production, 43 reduced proliferation of immunosuppressive regulatory T cells, 43 and a direct stimulatory effect on effector T lymphocytes. 44 In these studies, the enhanced anti-tumor immune response brought about by CDK4/6 inhibition was leveraged by the addition of immune checkpoint blockade, targeting pathways such as the programmed cell death protein-1 (PD-1) axis, and combined CDK4/6–PD1 blockade resulted in a synergistic effect on tumor growth. In one study, similar results were obtained in vivo when CDK4/6 inhibitors were combined with PI3K inhibitors and dual immune checkpoint blockade (inhibiting PD-1 and CTLA-4). 56
These results are intriguing, and are being actively followed up in several laboratories, including our own. Indeed, the promise of immunotherapy is the induction of durable responses in patients with advanced cancer, and if CDK4/6–immunotherapy combinations were to achieve this in breast cancer patients, it would be a very exciting development. It is important to note, however, that the early data described all come from mouse models. The extent to which these models reflect the biology of human cancer (and in particular ER-positive breast cancer, which typically shows little immune infiltrate) is unclear, and thus it remains to be seen whether this approach is valuable in patients. Preliminary results from the phase Ib JPCE study of abemaciclib plus pembrolizumab for patients with HR-positive, HER2-negative metastatic breast cancer were recently presented. At the 16-week interim analysis, there were no new safety signals and a confirmed ORR of 14.3% was observed. 60 Definitive data on the potential benefits of this strategy will only come from mature data and larger, randomized trials.
Continuing CDK4/6 inhibition beyond progression
One outstanding question is whether there is any role for the continuation of CDK4/6 inhibitors beyond progression on these agents. There are few preclinical data available to address this issue to date. It has been reported that CDK4/6 inhibitor-resistant cells might retain endocrine sensitivity, and anecdotes about cross-resistance between the three CDK4/6 inhibitors have been mixed. 61 This question of CDK4/6 inhibitor use beyond progression is being addressed in several clinical trials (see Table 5), some of them maintaining the CDK4/6 inhibitor while changing the endocrine agent and others changing the CDK4/6 inhibitor used.
Ongoing trials evaluating continuing CDK4/6 inhibition beyond progression in advanced ER-positive HER2-negative breast cancer (ClinicalTrials.gov January 2018).

CBR, clinical benefit rate; DLT, dose-limiting toxicities; PFS, progression-free survival.
Potential biomarkers of response and resistance to CDK4/6 inhibition
Clinical predictors
Currently, exploratory analyses of trials with CDK4/6 inhibitors have not identified a clinical subgroup of ER-positive patients that did not benefit from the addition of the CDK 4/6 inhibitors to endocrine therapy.7–10 A combined analysis of the MONARCH 2 and MONARCH 3 studies was recently presented, examining the impact of various clinical parameters (tumor grade, presence of liver metastases, PR status, treatment-free interval) on relative benefit from abemaciclib therapy. Although there was a suggestion that patients in the MONARCH 3 study with an (adjuvant) treatment-free interval of more than 3 years might benefit less from abemaciclib, this is exploratory data at best and should not be used to guide practice. 62
Molecular biomarkers
Despite available knowledge of the CDK4/6 pathway, attempts to identify molecular biomarkers that predict response or resistance to CDK4/6 inhibitors in human breast cancers have failed to identify clear candidates. Putative biomarkers are discussed in detail elsewhere (see the work of Garrido-Castro and colleagues 63 ), and are summarized briefly here.
RB expression
RB has an indispensable role in mediating anti-tumor responses to CDK4/6 inhibitors.7,32,64 As such, one would expect that tumors lacking functional RB are unlikely to respond to CDK4/6 inhibition. In support of this notion, clinical reports are emerging of tumors which acquired resistance to CDK4/6 inhibitors that show new polyclonal mutations in RB1 that are predicted to confer loss of function. 65 Despite this, analysis of large randomized trials has not revealed a clear association between levels of RB (as measured either by immunohistochemistry or gene expression) and benefit from CDK4/6 inhibitors.66–68 The reasons for this remain elusive, but may relate to the fact that neither of these assays adequately reflects RB functionality within a tumor. 69
Alterations in cyclin D1 or p16INK4A
Biologists have hypothesized that cancers bearing amplification of CCND1 should, by virtue of their high cyclin D1 protein levels, be more dependent on the CDK4/6 pathway and hence more vulnerable to CDK4/6 inhibition. Interestingly, analysis of patient samples has not shown this to be the case: the PALOMA-1 study suggested that CCND1 amplification does not predict benefit from palbociclib, 8 and PALOMA-2 samples similarly failed to show an association at the protein level. 66 Moreover, despite in vitro data suggesting that low expression of p16 would predict CDK4/6 inhibitor sensitivity, this has recently come into question and has not been recapitulated in clinical trials.7,8,17,66
Other gene expression profiles
Recently, gene expression analysis of the PALOMA-267 and PALOMA-368 tumor samples was performed, aiming to identify variables predictive of palbociclib benefit. One intriguing result from PALOMA-3 was that higher expression of the CCNE1 gene was associated with relative resistance to palbociclib [median PFS in patients with CCNE1 expression below median was 14.1 versus 4.8 months (HR 0.32 palbociclib versus placebo); median PFS in patients with CCNE1 expression above median 7.6 versus 4.0 months (HR 0.85 palbociclib versus placebo); interaction p = 0.0024]. 68 These data support a potential role of targeting CDK2 to subvert palbociclib resistance. However, this result was not upheld in analysis of the larger, first-line PALOMA-2 study, and clearly requires validation in other trial cohorts. 67 Finally, regarding intrinsic subtype, both luminal A and luminal B tumors derived benefit from palbociclib in both the PALOMA-2 and PALOMA-3 studies.67,68
Mutational profiles
Analysis of the PALOMA-3 trial has suggested that neither tumor mutations in PIK3CA nor ESR1 are associated with reduced benefit from palbociclib. 70 More recently, analysis of circulating cell-free DNA samples from patients on the MONALEESA-2 study showed negative prognostic implications of PIK3CA and TP53 mutations in patients with advanced ER-positive breast cancer, but neither was predictive of benefit from CDK4/6 inhibition. 71
Conclusions
CDK4/6 inhibitors have well and truly entered the treatment landscape for advanced ER-positive breast cancer, and in combination with hormonal treatment are now a standard first-line treatment option for both pre- and postmenopausal women. Importantly, the ORRs of over 50% seen in all first-line trials makes CDK4/6 inhibitor plus endocrine therapy regimens a suitable option over chemotherapy in most cases. In patients previously treated with endocrine therapy who are CDK4/6 inhibitor naïve, CDK4/6 inhibitors are also becoming a standard option. Furthermore, in more heavily pretreated patients, abemaciclib monotherapy can also be considered. Perhaps the most outstanding questions for patients with advanced ER-positive disease are whether CDK4/6 inhibitors will prolong their OS, and if there is any role in continuing treatment beyond progression. These answers will become clear as more trial data are presented in coming years.
As we have discussed, there is also a strong preclinical rationale for testing CDK4/6 inhibitors in other breast cancer subtypes (especially HER2-positive disease) and for evaluating novel CDK4/6 inhibitor combinations. As our understanding of the molecular mechanisms by which these drugs act improves, we are likely to be able to ask more refined questions, ultimately identifying optimal combinations that will benefit the majority of breast cancer patients, while sparing unnecessary toxicity and costs.
Footnotes
Acknowledgements
We thank Kaitlyn T. Bifolck for her editorial support to this work.
Funding
S. Goel is supported by a Career Development Award from the DF/HCC SPORE in Breast Cancer [National Institutes of Health (NIH) 2015 P50 CA] and a Young Investigator Grant from the Breast Cancer Alliance. S. Pernas is supported by grants from Fundación AECC (Asociación Española Contra el Cáncer) and the Spanish Society of Medical Oncology (SEOM).
Conflict of interest statement
S. Pernas has received honoraria for talks and travel grants from Roche, outside of the submitted work, and has served on advisory boards for Polyphor. S. Goel performs laboratory research sponsored by Eli Lilly and serves as a paid adviser to Eli Lilly. S. Tolaney receives institutional research funding from Eli Lilly, Pfizer, Novartis, Exelixis, Eisai, Merck, Bristol Meyers Squibb, AstraZeneca, Nektar. S. Tolaney has also served on advisory boards for Eli Lilly, Pfizer, Novartis, Eisai, Merck, AstraZeneca, Nektar, and Puma. E Winer has been a consultant to Roche and Eli Lilly.