
Abstract
Background:
Epithelial ovarian cancer (EOC) is characterized by exacerbated angiogenesis regulated by proangiogenic and growth factors. Nerve growth factor (NGF) is overexpressed in EOC where it promotes proliferation as well as survival and is considered a proangiogenic factor. Metformin, a drug commonly used in the treatment of diabetes, is attributed to antineoplastic effects, but the underlying mechanisms remain unknown. Given that current therapies yield modest results in EOC patients, the aim of this study was to determine the effects of metformin on NGF-enhanced proliferation of EOC cells and the angiogenic potential of endothelial cells.
Methods:
A2780 (EOC), HOSE (human ovarian surface epithelial) and EA.hy926 (endothelial) cells were treated with NGF and metformin. Cell viability, cell proliferation and cell cycle were evaluated in all three cell lines, and the angiogenic potential in endothelial EA.hy926 cells.
Results:
NGF enhanced cell proliferation in A2780, HOSE and EA.hy926 cells (p < 0.05), while metformin treatment decreased cell proliferation in A2780 and EA.hy926 cells (p < 0.05). Moreover, the NGF-enhanced angiogenic score in EA.hy926 cells was prevented by metformin (p < 0.05).
Conclusions:
Given that NGF plays a significant role in EOC progression, our current findings suggest that metformin holds considerable promise as an adjuvant treatment in ovarian cancer.
Introduction
Ovarian cancer is one of the most aggressive types of cancer with poor prognosis and represents the third most important cause of death among gynecological cancers. 1 Approximately 80% of ovarian cancer cases are considered to be the serous epithelial ovarian cancer (EOC) type, 2 and are characterized by unusually high levels of angiogenesis. 3 Unfortunately, EOC is usually diagnosed at advanced stages, which translates into low survival rates,2,4,5 and current therapies are only moderately successful.6,7 Thus, studies are required to understand the molecular mechanisms governing the progression of this cancer in order to identify new therapeutic targets and treatments.
Neurotrophins and their receptors have been found in several nonneural tissues, including the ovary.8,9 One of the best characterized neurotrophins, nerve growth factor (NGF), participates in follicular development and ovulation. 10 Our research group has studied the role of NGF in EOC and our findings indicate that NGF interaction with the high affinity receptor, tropomyosin receptor kinase A (TrkA), promotes proliferation, survival and angiogenesis.11–13 Furthermore, NGF has been shown to act directly on tumor cells as well as indirectly on endothelial cells to promote angiogenesis. 14 Of note, activation of the TrkA receptor on endothelial cells induces vascular endothelial growth factor (VEGF) synthesis in EOC explants, a growth factor that promotes angiogenesis by acting on endothelial cells.11,14
Despite the demonstrated importance of NGF and TrkA in EOC progression, these molecules cannot be considered as therapeutic targets because they play important roles in several tissues, mainly in the development and maintenance of the central and peripheral nervous system.15,16 Therefore, the challenge is to identify new therapeutic alternatives with the capacity to selectively affect only cancer cells.
Given the anticancer properties attributed to metformin in several cancer models, including EOC, this drug has received considerable attention in the search for new drugs in EOC treatment. 17 Metformin is a biguanide, and its therapeutic indications include treatment for polycystic ovarian syndrome, gestational diabetes, type II diabetes mellitus, insulin resistance and metabolic syndrome.18–20 Observational studies indicate that metformin offers some degree of protection against cancer development in diabetic patients. 21 For instance, Bodmer and colleagues performed a case-control study with 1611 EOC patients comparing those that used metformin with patients that did not take this drug. The authors concluded that those who had taken metformin had a lower risk of developing ovarian cancer. 22 Kumar and colleagues also found in their case-control study an association between metformin use and an increase in overall survival in patients with ovarian cancer. 23 Similar results were found in a study by Romero and colleagues that concluded that diabetic women who take metformin and suffer from ovarian cancer have a higher survival rate and a lower risk of recurrence compared with diabetic women with ovarian cancer without metformin treatment. These protective effects of metformin are also observed in ovarian cancer patients without diabetes. 24 These findings highlight the necessity of studying the possible anticancer mechanisms of this drug in EOC.
The mechanism of action of metformin has been mostly studied at the hepatic level, where it inhibits mitochondrial complex I and activates AMP-activated protein kinase (AMPK). AMPK inhibits the PI3K-AKT-mTOR pathway, 25 inducing a metabolic switch that favors catabolic processes. However, it is important to mention that several AMPK-independent effects have been discovered, meaning that metformin has pleiotropic effects. 26 In addition, in vitro studies showed that metformin can inhibit the MAPK/ERK signaling pathway,27,28 a relevant signaling pathway for cell survival and proliferation.29,30 Interestingly, after the interaction of NGF with TrkA, the PI3K-AKT and MAPK/ERK pathways are activated.13,31 Therefore, we hypothesized that metformin may be acting in EOC by inhibiting the effects of the NGF/TrkA system.
Considering that NGF levels increase in EOC 12 and that NGF stimulates cell proliferation and angiogenesis in EOC explants,12,13 we sought to determine here whether metformin treatment alters NGF-induced processes in EOC and endothelial cells. To that end, in vitro experiments were performed on cell lines derived from the ovarian surface epithelium and on a human endothelial cell line. All cell lines were treated with metformin in order to determine if this drug interferes with NGF-induced proliferation and angiogenesis.
Materials and methods
Cell lines and materials
A total of three cell lines were used: A2780 cells (a human ovarian cancer cell line with epithelial morphology, originated from a primary ovarian tumor), HOSE cells (human ovarian surface epithelial cells from a menopausal woman, immortalized by SV40-Tag), and EA.hy926 cells (human endothelial cells obtained from the immortalization of human umbilical vein endothelial cells). Cells were routinely checked for mycoplasma contamination. A2780 and EA.hy926 cells were obtained from the American Type Culture Collection and HOSE cells were donated by Dr Davie Munroe (NCI, NIH, USA).
Cells were grown in phenol red-free Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 medium (Sigma-Aldrich Co. St. Louis, MO, USA) supplemented with 2% fetal bovine serum (Hyclone™ Thermo Fisher Scientific, Massachusetts, USA), and stimulated with NGF (Sigma-Aldrich Co.) or metformin chlorhydrate (Sigma-Aldrich Co.) following two different experimental protocols: (1) cell cycle was evaluated with metformin treatment for 48 h plus NGF stimulation during the last 6 h; (2) cell viability and cell number were measured after 48 h of co-stimulation with NGF and metformin. This design was used because NGF acts in short frames of time, and the doubling time for A2780 cells is short (around 18 h). 32 The TrkA receptor-specific inhibitor GW441756 (Tocris, Bristol, UK) was used at a final concentration of 20 nM and the NGF-neutralizing antibody at a final concentration of 5 μg/ml (ab6199, Abcam, Cambridge, UK).
Viability and cell counting assays
In 96-well plates, 5000 cells were cultured and stimulated with 25, 50 or 100 mg/ml of NGF or metformin at concentrations of 0.5 mM, 1 mM, 5 mM and 10 mM for 48 h. Afterwards, cell viability was evaluated using the cell cytotoxicity assay commercial kit (Abcam), according to the manufacturer instructions. In parallel experiments, cells were stimulated as described above and counted after trypan blue staining (0.4%) in a Neubauer chamber and using the LUNA system (Logos Biosystems, Anyang, South Korea) following staining with acridine orange and propidium iodide (Logos Biosystems), to visualize live and dead cells by fluorescence.
Ki 67 immunocytochemistry
Cells (10,000) were grown on 12 mm round coverslips and stimulated with 10 mM metformin for 48 h, 100 ng/ml of NGF for 6 h or metformin for 48 h plus NGF in the last 6 h. Once stimulation experiments were completed, cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% triton X-100 and incubated for 15 min at room temperature with 3% hydrogen peroxide. Ki67 was detected with a primary anti-Ki67 antibody (sc-23900, Santa Cruz Biotechnology, Texas, USA) diluted 1:100 for 1 h at 37°C. Afterwards, cells were washed and incubated with a horse radish peroxidase-coupled antimouse secondary antibody (KPL 074-1806, SeraCare, Milford, MS, USA) diluted 1:300 for 45 min at 37°C. To detect bound antibodies, cells were washed and then incubated with 3.3′-diaminobenzidine (DAB) (DakoCytomation, Inc., CA, USA) as a substrate. Slides were evaluated using an optic microscope (Olympus Corporation, Tokyo, Japan) and images were obtained with a Micro-Publisher 3.3 RTV camera (Q Imaging, Surrey, BC, Canada). Finally, immunodetection was evaluated by obtaining the integrated optical density with the computer software Image Pro Plus 6.2 (Media Cybernetics Inc., Silver Spring, MD, USA).
Flow cytometry analysis of cell cycle and cell death
Cells (200,000) were cultured in six-well plates and stimulated with 10 mM metformin for 48 h, 100 ng/ml of NGF for 6 h or metformin for 48 h plus NGF during the last 6 h. Supernatants and cells were collected and centrifuged at 100 g for 5 min at 4°C. Afterwards, cells were permeabilized in methanol at −20°C, centrifuged and resuspended in FACS phosphate-buffered saline 1×. Finally, cells were treated with ribonuclease A (Sigma-Aldrich Co.) at a final concentration of 100 μg/ml for 1 h at 37°C. Cells were then dispersed in a 1 ml tuberculin syringe, and propidium iodide was added (Invitrogen, California, USA) at a final concentration of 10 μg/ml. Cell cycle stages or cell death were analyzed by flow cytometry in the BD FACS Canto A equipment (BD Biosciences, NJ, USA). In every sample, 10,000 events were measured and data were analyzed with the De Novo FCS Express v6.03.0011 software.
Matrigel vasculogenesis assays (angiogenic score)
EA.hy926 cells were used for this essay. Cells were serum-deprived for 24 h, and trypsinized in order to culture 10,000 cells in 500 μl of phenol red-free and serum-free DMEM/Ham’s F-12 medium. Cells were then stimulated with 50 ng/ml or 100 ng/ml of NGF or metformin with concentrations of 1, 5 and 10 mM. Afterwards, cells were plated in 24-well plates covered by 150 μl of growth factor-free and phenol red-free Matrigel (Corning, New York, USA) for 8 h. Then, cells were photographed and the angiogenic score 33 was measured for each experimental condition, according to this formula:

For each experiment, eight images were obtained and each of them was analyzed individually with the Fiji ImageJ and the cell counter plugin (https://imagej.nih.gov/ij/plugins/cell-counter.html). Also, images were processed with the angiogenesis analyzer plugin (ImageJ, https://imagej.nih.gov/ij/macros/toolsets), which allowed for the measurement of several parameters, including the number of polygonal structures (meshes) and the number of multicellular unions (junctions), as shown in Supplemental Figure 5.
Migration assay
EA.hy926 cells were serum-deprived for 24 h, and stimulated with NGF (100 ng/ml) and metformin (10 mM) for 6 h. Then, supernatants were removed, cells were trypsinized and 100,000 cells resuspended in the same conditioned supernatant and were added to the upper chamber of 6.5 mm Transwell® with 8.0 µm pore polycarbonate membrane insert, (Corning) coated on the lower surface with fibronectin (Gibco™ Thermo Fisher Scientific). Cells were allowed to migrate for 2 h at 37°C. After this, EA.hy926 cells were stained overnight with crystal violet and cells that did not cross the membrane were discarded with a cotton swab, while cells attached to the lower membrane surface were counted. Inserts were photographed (eight pictures in each experimental condition) and analyzed using Fiji ImageJ (cell counter plugin).
Statistical analysis
Data were expressed as percentage ± standard error of mean. Data were analyzed with the nonparametric Kruskal–Wallis test and Dunn posttest, or with a Mann–Whitney test. All data were plotted as percentage of fold change with respect to the basal condition (without treatment).
Results
NGF increases proliferation of HOSE, A2780 and EA.hy926 cells
In order to determine whether NGF alters cell proliferation of ovarian and epithelial cells, a dose–response curve for NGF was performed and cell viability and the number of HOSE, A2780 and EA.hy926 cells were assessed. As shown in Supplemental Figure 1, 50 ng/ml of NGF induced a significant increase in A2780 and HOSE cell viability (p < 0.05); while 100 ng/ml promoted a significant increase in the viability of all three cell lines [p < 0.05; Supplemental Figure 1(a–c)]. Additionally, both NGF concentrations increased the number of A2780, HOSE and EA.hy926 cells beyond baseline values after 48 h of stimulation [Figure 1(a–c)] and this effect is blocked by a specific TrkA inhibitor or anti-NGF antibody (data not shown). Also, with 100 ng/ml of NGF no change in the number of cells undergoing cell death was observed [Figure 1(d–f)].

NGF increases the proliferation of A2780, HOSE and EA.hy926 cells.
To complement the previous results, the cell cycle marker Ki 67 was evaluated in A2780, HOSE and EA.hy926 cells. NGF increased Ki 67 immunostaining to 93.6% in A2780 cells (p < 0.01), 63.3% in HOSE cells (p < 0.05) and 50% in EA.hy926 cells (p < 0.05) [Figure 1(g–i)]. Importantly, in the presence of GW441756 (GW), a specific TrkA inhibitor, or a neutralizing antibody against NGF (Ab), NGF-induced effects on Ki 67 immunostaining were reversed [Figure 1(g–i)]. These pharmacological and immunological approaches confirm that NGF increases Ki 67 immunostaining through its interaction with the TrkA receptor.
Additionally, the percentage of cells in each stage of the cell cycle was determined by flow cytometry. In A2780 cells, NGF significantly increased the percentage of cells in the G2/M phase of the cell cycle (p < 0.05). Alternatively, a trend towards a decrease in the percentage of cells in G0/G1 phases of the cell cycle (p = 0.0519) was detectable [Figure 1(j) and Supplemental Table 1]. For HOSE cells, NGF stimulation increased the percentage of cells in the G2/M phase (p < 0.05) while decreasing those in the G0/G1 phase of the cell cycle (p < 0.05) [Figure 1(k), Supplemental Table 1]. Also for the EA.hy926 cells, NGF significantly increased the percentage of cells in the G2/M phases (p < 0.05) [Figure 1(l), Supplemental Table 1]. Moreover, the effects of NGF observed in all lines were blocked by the TrkA inhibitor (GW) and the NGF-neutralizing antibody (p < 0.05 and p < 0.01) [Figure 1(j–l) and Supplemental Table 1]. Taken together, these results show that NGF increases A2780, HOSE and EA.hy926 cell proliferation via a TrkA-dependent mechanism.
Metformin decreases NGF-enhanced proliferation of HOSE, A2780 and EA.hy926 cells
The effect of different concentrations of metformin (0.5, 1, 5 and 10 mM) on the viability of A2780, HOSE cells was subsequently evaluated. In A2780 cells, concentrations of 5 mM or 10 mM metformin induced a statistically significant decrease in cell viability after 48 h of treatment (p < 0.001) [Supplemental Figure 2(a)], while all concentrations of metformin decrease cell viability of EA.hy926 cells [p < 0.05 and p < 0.001; Supplemental Figure 2(c)]. For HOSE cells, however, no changes in cell viability were observed after metformin treatment; on the contrary, low concentrations of metformin increased the viability of these noncancer cells [p < 0.05; Supplemental Figure 2(b)]. Moreover, metformin (10 mM) significantly decreased the number of A2780 cells by 55.9% with respect to baseline (p < 0.01) [Figure 2(a)], and also decreased by at least 30% the number of EA.hy926 cells when used at the concentrations of 5 mM and 10 mM [p < 0.05 and p < 0.01; Figure 2(c)]. The number of HOSE cells was not altered by metformin [Figure 2(b)]. Our results also show that metformin did not alter cell death in any of the three cell lines studied here [Figure 2(d–f)].

Metformin decreases proliferation of A2780 and EA.hy926 cells.
Notably, treatment with 10 mM metformin decreased Ki 67 immunostaining in A2780 and EA.hy926 cells (p < 0.01 and p < 0.05), without inducing significant changes in HOSE cells [Figure 2(g, h and i)]. Furthermore, in A2780 cells, metformin tended to increase the percentage of cells in the G0/G1 phase (p = 0.0563), while decreasing by 53% the cells in S phase [p < 0.05; Figure 1(j)]. In a similar manner, metformin significantly increased the percentage of HOSE cells in the G0/G1 phase (p < 0.05) and decreased the percentage of cells in the S phase [p < 0.01; Figure 1(k)], without changing the percentage of cells found in the G2/M phase. In EA.hy926 cells, metformin significantly reduced the percentage of cells in the G2/M phase [p < 0.05; Figure 1(l)].
Taken together, these results indicate that metformin reduces the proliferation of A2780 and EA.hy926 cells, without having noticeable effects on HOSE cells.
Metformin reduces NGF-enhanced viability and increases the number of HOSE, A2780 and EA.hy926 cells
To determine whether metformin can prevent NGF-enhanced viability and the number of A2780, HOSE and EA.hy926 cells, cells were co-treated with NGF (100 ng/ml) and metformin (1 mM and 10 mM) for 48 h. When A2780 and EA.hy926 cells were co-stimulated with NGF (100 ng/ml) and metformin (1 mm), the increase in cell viability induced by NGF was not significantly affected [Figure 3(a and c) and Supplemental Figure 3]; however, co-treatment with 5 mM and 10 mM metformin did prevent NGF-enhanced cell viability (p < 0.01) [Supplemental Figure 3; Figure 3(d and f)]. In HOSE cells, the co-treatment with NGF and metformin (1 mM and 5 mM) did not significantly affect NGF-enhanced cell viability [Supplemental Figure 3 and Figure 3(b–e)]; however, when comparing NGF and NGF + metformin (10 mM) treatments, we did observe a decrease in the NGF-induced effect by metformin (p < 0.05).

Metformin treatment precludes NGF-enhanced viability and reduces the number of A2780, HOSE and EA.hy926 cells.
Similar results were obtained in cell counting experiments, which revealed that 10 mM metformin blocked the NGF-enhanced number of A2780 and EA.hy926 cells (p < 0.05). For the metformin concentrations 0.5 mM (Supplemental Figure 3) and 1 mM [Figure 3(g, i, j and l)] the reduction was not as significant. HOSE cells show the same behavior we previously described for cell viability, in that metformin did not reduce NGF-induced effects in a statistically significant manner [Figure 3(h and k) and Supplemental Figure 3].
Metformin prevents the NGF-enhanced proliferation and cell cycle progression of HOSE, A2780 and EA.hy926 cells
Next, we determined the effect of metformin (10 mM) and NGF (100 ng/ml) co-treatment on the presence of cell proliferation marker Ki 67 and on cell cycle progression of A2780, HOSE and EA.hy926 cells. Metformin treatment prevented NGF-enhanced Ki 67 immunostaining [Figure 4(a–d)] in all three cell lines (p < 0.001). In A2780 and HOSE cells, metformin significantly decreased the number of cells in G0/G1 (p < 0.05 and p < 0.01); and increased those in the S phase of the cell cycle induced by NGF [p < 0.05 and p < 0.01; Figure 4(e and g), Supplemental Table 1]. In EA.hy926 cells, on the other hand, metformin prevented the NGF-induced decrease in cells in the G0/G1 phase (p < 0.05) [Figure 4(f), Supplemental Table 1].

Metformin treatment reduces NGF-enhanced Ki-67 immunodetection and cell cycle progression in A2780, HOSE and EA.hy926 cells.
Additionally, we also evaluated cell death in all three lines. Our findings show that NGF and metformin did not induce significant changes regarding the basal condition, although NGF and metformin co-treatment increased the number of A2780 cells undergoing cell death compared with cells treated with NGF alone [p < 0.05; Figure 4(h–j)].
NGF increases while metformin decreases the angiogenic score and migration of EA.hy926 cells
To determine whether metformin and NGF modulated cell migration and differentiation, both relevant processes for the angiogenic potential of EA.hy926 cells, tube formation and migration were evaluated in Matrigel and migration assays, respectively.
Following stimulation of EA.hy926 cells with 100 ng/ml of NGF for 8 h, the angiogenic score increased by 52.7%, the average number of junction structures (multicellular joints) from 5.9 to 13.9 and the average number of polygonal structures, referred to as ‘meshes’, from 1.3 to 3.3 [Figure 5(a–d)]. While these effects are statistically significant (p < 0.05), they are less pronounced than the effect of VEGF (Supplemental Figure 5). Furthermore, NGF (100 ng/ml) increased the migration of EA.hy926 cells by 75.5% [p < 0.001; Figure 6(e and f)]. On the other hand, metformin induced opposite effects: 5 mM and 10 mM metformin decreased the number of junctions (p < 0.05), and significantly reduced by more than 30% the angiogenic score in the same time frame [p < 0.05; Figure 5(e–h)] as well as the migration of these cells by 48.1% (p < 0.001) compared with control group [Figure 6(e and f)].

NGF enhanced, while metformin reduced, the angiogenic score of EA.hy926 cells.
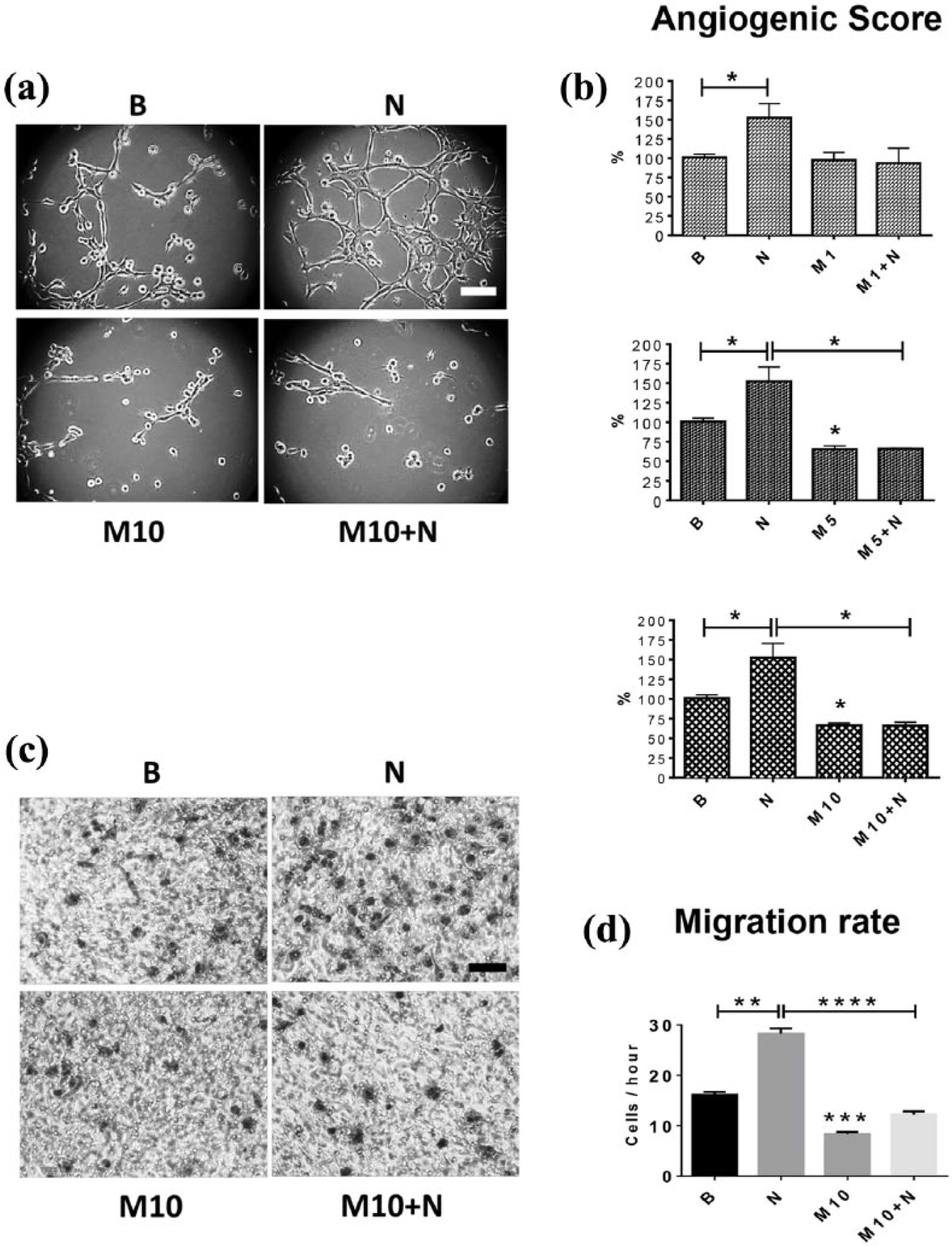
Metformin reduces the NGF-enhanced angiogenic score and migration of EA.hy926 cells.
Metformin prevents the NGF-induced increase in the angiogenic score and migration of EA.hy926 cells
Finally, we evaluated the effect of co-treatment with NGF and metformin on the angiogenic score and migration of EA.hy926 cells. Interestingly, 1 mM metformin did not reduce the NGF-induced increase in the angiogenic score of these cells (p = 0.2286). However, co-treatment with NGF and 5 mM or 10 mM metformin completely prevented the NGF-induced increase in the angiogenic score of EA.hy926 cells [p < 0.05; Figure 6(a–d)]. Likewise, metformin (10 mM) blocked the NGF-induced increase in the migration of EA.hy926 cells (p < 0.0001).
Discussion
Little information concerning the effects of metformin on growth factor signaling, other than insulin, is currently available. Given that NGF levels are elevated in EOC, 12 and that NGF was shown to promote cell proliferation and angiogenesis in EOC explants,13,34 our studies sought to determine whether metformin altered such NGF-induced events in endothelial cells and EOC cells. We observed that NGF increased proliferation of the A2780 ovarian carcinoma cells, as well as of HOSE cells, a noncarcinogenic ovarian epithelial cell line. Metformin, on the other hand, blocked EOC cell proliferation induced by NGF. Also, NGF was shown to favor angiogenic behavior in vitro of EA.hy926 cells, while metformin blocked the increase in the angiogenic score and migration triggered by NGF stimulation. Considering that the doubling time of EA.hy926 is 25.3 h, 33 one may assume that in 2 h only 8% of cells should have divided, which is substantially inferior to the decrease by 42.6% in migration after 10 mM metformin treatment (Figure 6). Bearing this in mind, the time chosen to evaluate EA.hy926 cell migration (2 h) allows us to exclude the possibility that differences in migration are due to alterations in proliferation. Taken together, our observations indicate that metformin blocks NGF-induced effects in EOC and endothelial cells.
Neurotrophins and their receptors play an important role in the normal ovarian function. 10 Moreover, NGF and TrkA are overexpressed in EOC, and are thought to contribute to disease progression. 12 In EOC explants, NGF activates signaling pathways related to cell survival and proliferation; as well as induces the expression of protumoral proteins, for example cMYC transcription factor and BCL-2. 13 Here, using cell lines we observed that NGF promoted proliferation of not only A2780 cells, but also of noncarcinogenic HOSE cells. The experiments with the pharmacological inhibitor TrkA receptor GW441756 and with a NGF-neutralizing antibody indicate that the increase in cell proliferation induced by NGF is a specific effect of this neurotrophin that is dependent on interaction with the TrkA receptor (Figure 1).
NGF has been proposed to trigger angiogenic effects both via direct and indirect mechanisms on the tumor and endothelial cells, respectively, in EOC. 14 Experiments performed on EOC explants and A2780 cells show that NGF increases VEGF levels,11,35,36 one of the best characterized proangiogenic factors. In addition, endothelial cells from tumor blood vessels express TrkA, as do EA.hy926 cells; 12 therefore, both cell types have the ability to respond to NGF. Conditioned media from A2780 cells, previously stimulated with NGF, increase EA.hy926 cell differentiation, migration and viability and this effect is reversed by the use of tyrosine kinase inhibitor K252a or a neutralizing antibody against NGF. 12 Here, we found that these cells respond to direct stimulation with NGF by increasing their angiogenic capacity. As mentioned, angiogenesis is one of the distinguishing characteristics of EOC. Since angiogenesis contributes to progression and dissemination of this cancer, therapies targeting angiogenesis have been developed as a strategy to treat EOC. An antibody against VEGF (bevacizumab), 37 for instance, is currently in use for patients who suffer from EOC at advanced stages. Here it is important to note that while bevacizumab produces a significant improvement of progression-free survival, it does not improve overall average survival. 38 This may be explained by VEGF-independent revascularization, which is thought to allow the tumor to continue growing, because other angiogenic factors contribute to tumor adaptation and angiogenesis. 39 It is intriguing to speculate that NGF may be one such factor.
Despite the potential relevance of NGF and the TrkA receptor in EOC, neither constitute suitable therapeutic targets, because they fulfill important functions in other tissues.15,16 As previously mentioned, NGF interaction with TrkA activates several signaling pathways, including PI3K/AKT and MAPK/ERK pathways, 31 which induce cell survival and proliferation. Interestingly, the antidiabetic drug metformin, considered safe and cost-effective, has been on the market for years and is known to modulate these signaling pathways precisely. Moreover, several reports have associated metformin use in diabetic patients with a reduction in cancer incidence and mortality.21,28 For EOC, only a few studies are available showing that metformin use increases survival and reduces the risk of cancer recurrence.22–24 Moreover, metformin has been used on EOC cells at concentrations ranging from a few μm to hundreds of mm.7,40–43 Metformin plasma concentrations do not reflect tissue concentrations, because this drug has a high apparent volume of distribution,44,45 meaning that it accumulates in tissues. Studies performed on rats have shown that metformin accumulates in abdominal organs, 46 mainly due to the number of cationic transporters found at the cellular level. 47 These transporters can also be found in the ovary, 48 and it has been described that they are relevant in the uptake of antineoplastic drugs, altering the response to EOC treatment. 49 Taking this into account, we considered it likely that metformin might also accumulate in the ovary beyond the plasma level, to reach on the order of mM concentrations. For these reasons, the effect of metformin was tested in cell lines in the range from 0.5 mm to 10 mm. In these experiments, metformin reduced the proliferation on A2780 cancer cells and on EA.hy926 endothelial cells (Figure 2). These findings support the idea that metformin has both cytostatic and antiangiogenic effects, which represent traits likely to be beneficial against EOC. Importantly, the effects of metformin cancer cells were not seen in the noncancer cell line, HOSE. Particularly this latter observation makes metformin an attractive therapeutic option for EOC treatment that should have few detrimental side effects in normal cells.
Metformin is a known activator of AMP-dependent kinase (AMPK) in hepatocytes, 50 and the role of this kinase is controversial in cancer. Studies have shown that AMPK activation reduces cell proliferation, which is essential for tumor growth. However, AMPK also participates in the activation of autophagy and adaptation to a lack of nutrients in the cell, thereby promoting survival.32,51 Indeed, reports are available showing that the antitumoral effects of metformin may be AMPK-dependent or independent. For example, in ovarian cancer cells OVCAR3, metformin treatment (1 mM, 72 h) activates AMPK, increases protein acetylation, and alters gene expression and that these effects are dependent on AMPK activity. 52 Alternatively, in breast cancer cells, metformin-induced activation of AMPK decreases cell proliferation by arresting cells in G1 phase. 53 Nevertheless, there is evidence that in the absence of AMPK, metformin can still trigger effects. For instance, in AMPK-null mouse embryonic fibroblasts (MEFs) as well as AMPK-silenced ovarian cancer cells, metformin decreased cell proliferation in a manner similar to control cells expressing AMPK. 54 Similarly, MEFs defective in LKB1-AMPK signaling remain sensitive to the cytostatic effects of metformin. 55 Moreover, in HeLa cells, NGF promotes cell viability under glucose starvation conditions, in a manner dependent on AMPK activation. 56 In conjunction, these observations indicate that metformin’s effects on the NGF/TrkA signaling may not require AMPK activation; however, additional experiments will be required to clarify this point. On the other hand, metformin has been reported to regulate the anti-inflammatory response. For example, this drug reduces lipopolysaccharide-induced interleukin (IL)-1β in macrophages, 57 decreases bladder cancer progression by inhibiting COX-2/PGE2 58 and also inhibits IL-8 induction in colon cancer cells stimulated with tumor necrosis factor (TNF)α by decreasing nuclear factor (NF)-κβ DNA-binding activity. 59 Because inflammation plays a fundamental role in cancer, metformin might potentially be inhibiting NGF/TrkA-mediated inflammatory responses.
To the best of our knowledge, no reports appear to be available characterizing the possible association between NGF/TrkA signaling and metformin in cancer. Thus, the present study evaluated whether this drug had an effect on the NGF/TrkA system, which is highly activated in EOC. 12 The results revealed that metformin (10 mM) completely prevented the increase in A2780 cell proliferation after NGF stimulation (Figures 3 and 4), but the effect was essentially not observed when NGF was combined with lower concentrations of metformin (0.5 mM and 1 mM) (Figure 3 and Supplemental Figure 3). Likewise, in EA.hy926 endothelial cells, metformin prevented the increase in proliferation and angiogenic capacity induced by NGF (Figures 3, 4 and 6). Of note here is that cell viability assays that measure mitochondrial activity are wildly employed in studies with metformin; however, metformin is known to inhibit the mitochondrial complex I.49,50,60,61 Thus, such studies do not truly evaluate viability or proliferation. For these reasons, we used additional assays that evaluated cell numbers (trypan blue assay) or proliferative nuclei (Ki67 staining) and essentially obtained similar results.
A potential limitation of this study is that only the effects on the hyperactivated NGF/TrkA system in EOC were studied. However, other growth factors are known to promote tumor progression and angiogenesis, including the epidermal growth factor and the fibroblast growth factor. 39 However, it is important to highlight that these growth factors also activate the same downstream signaling pathways known to be relevant to NGF/TrkA signaling. Hence, if metformin inhibits these pathways downstream of NGF/TrkA, it is likely that metformin should also be effective in preventing signaling events triggered by these other growth factor receptors. Future studies will evaluate these attractive possibilities.
Conclusion
The results shown here indicate that NGF increases the proliferation of A2780, HOSE and EA.hy926 cells and the angiogenic potential of EA.hy926 cells. On the other hand, metformin decreases the proliferation of A2780 and EA.hy926 cells, without inducing significant changes in HOSE cells, while decreasing the angiogenic potential of EA.hy926 cells. Co-treatment experiments using NGF and metformin revealed that metformin prevents NGF-induced proliferation and proangiogenic effects in the cell lines studied here. Both these processes are important for the progression and dissemination of EOC. Thus, the tumor suppressor effects of metformin may, in part, be attributable to its ability to block the effects mediated by NGF and given the relevance of this signaling pathway in EOC, metformin should be considered as an adjuvant in therapeutic protocols for the treatment of this cancer.
Supplemental Material
Garrido_et_al._Supl_Material – Supplemental material for Metformin prevents nerve growth factor-dependent proliferative and proangiogenic effects in epithelial ovarian cancer cells and endothelial cells
Supplemental material, Garrido_et_al._Supl_Material for Metformin prevents nerve growth factor-dependent proliferative and proangiogenic effects in epithelial ovarian cancer cells and endothelial cells by Maritza P. Garrido, Carolina Vera, Margarita Vega, Andrew F.G. Quest and Carmen Romero in Therapeutic Advances in Medical Oncology
Footnotes
Author contributions
Carmen Romero, Andrew Quest and Maritza Garrido designed the experiments and analyzed the data; Maritza Garrido performed the experiments; and Maritza Garrido, Carolina Vera, Margarita Vega, Carmen Romero and Andrew Quest wrote the paper.
Funding
The studies reported here were supported by FONDECYT (grant number 1160139) to CRO, CONICYT-FONDAP (grant number 15130011), FONDECYT (grant numbers 1130250, 1170925) to AFGQ, and a CONICYT scholarship (number 21150360) to MG.
Conflict of interest statement
The authors declare that there is no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.