
Abstract
The disease course of BRAF (v-raf murine sarcoma viral oncogene homolog B1)-mutant melanoma has been drastically improved by the arrival of targeted therapies. NRAS (neuroblastoma RAS viral oncogene homolog)-mutated melanoma represents 15–25% of all metastatic melanoma patients. It currently does not have an approved targeted therapy. Metastatic patients receive immune-based therapies as first-line treatments, then cytotoxic chemotherapy like carboplatin/paclitaxel (C/P), dacarbazine (DTIC) or temozolomide (TMZ) as a second-line treatment. We will review current preclinical and clinical developments in NRAS-mutated melanoma, and analyze ongoing clinical trials that are evaluating the benefit of different targeted and immune-based therapies, either tested as single agents or in combination, in NRAS-mutant melanoma.
Introduction
Malignant melanoma represents less than 5% of all cutaneous malignancies but accounts for the majority of deaths from skin cancer. 1 Due to its increasing incidence in White populations, in the USA, it is now the fifth leading cancer in men and the seventh in women. 1
Patients diagnosed with localized melanoma at an early stage have a good chance of survival and are treated solely with surgery. 2 Treatment of advanced or metastatic disease is dependent on the genotype of the melanoma with four distinct genetic categories including BRAF (v-RAF murine sarcoma viral oncogene homolog B1) mutant, NRAS (neuroblastoma RAS viral oncogene homolog) mutant, NF1 mutant and triple negative mutant melanoma (or wild type; WT) which includes melanomas with GNAQ or KIT mutations. 3
The mitogen activated protein kinase (MAPK) cell signaling pathway, (also known as the RAS-RAF-MEK-ERK pathway) regulates cell growth, proliferation and differentiation in response to growth factors, cytokines and hormones and it is frequently altered in melanoma with 50% of metastatic cutaneous melanoma patients harboring a BRAF-activating mutation 4 and 20–30% of them harboring an NRAS-activating mutation. 5
The disease course of BRAF-mutant melanoma has been improved recently by the advent of targeted therapies, like BRAF inhibitors (BRAFi) (vemurafenib, dabrafenib, and encorafenib) that are used alone or in combination with MEK inhibitors (MEKis) (cobimetinib, trametinib, and binimetinib) and by the arrival of new immune-based therapies, that activate the immune system by targeting immune checkpoints (ipilimumab, nivolumab, pembrolizumab). 6
NRAS-mutated melanoma currently does not have an approved targeted therapy and metastatic patients receive immune-based therapies as first-line treatment, then cytotoxic chemotherapy like carboplatin/paclitaxel (C/P), dacarbazine (DTIC) or temozolomide (TMZ) as a second-line treatment. 6 We will review current preclinical and clinical developments in NRAS-mutated melanoma, and analyze ongoing clinical trials that are evaluating the benefit of different targeted and immune-based therapies, tested as single agents or in combination, in NRAS-mutant melanoma.
Characteristics of NRAS melanoma
Three RAS family genes are known to be mutated in 20% of human cancer: NRAS, HRAS (Harvey Rat sarcoma virus) and KRAS (Kirsten Rat sarcoma virus). 7 RAS proteins are small plasma membrane-associated guanosine 5’-triphosphate (GTP)-binding proteins that regulate cell growth by transmitting the signal from receptor tyrosine kinases (RTKs) at the cell surface to transcription factors and cell cycle proteins in the nucleus 7 (Figure 1a). Oncogenic RAS proteins also have a role in tumor cell metabolism, microenvironment remodeling, and tumoral immune response evasion. 8
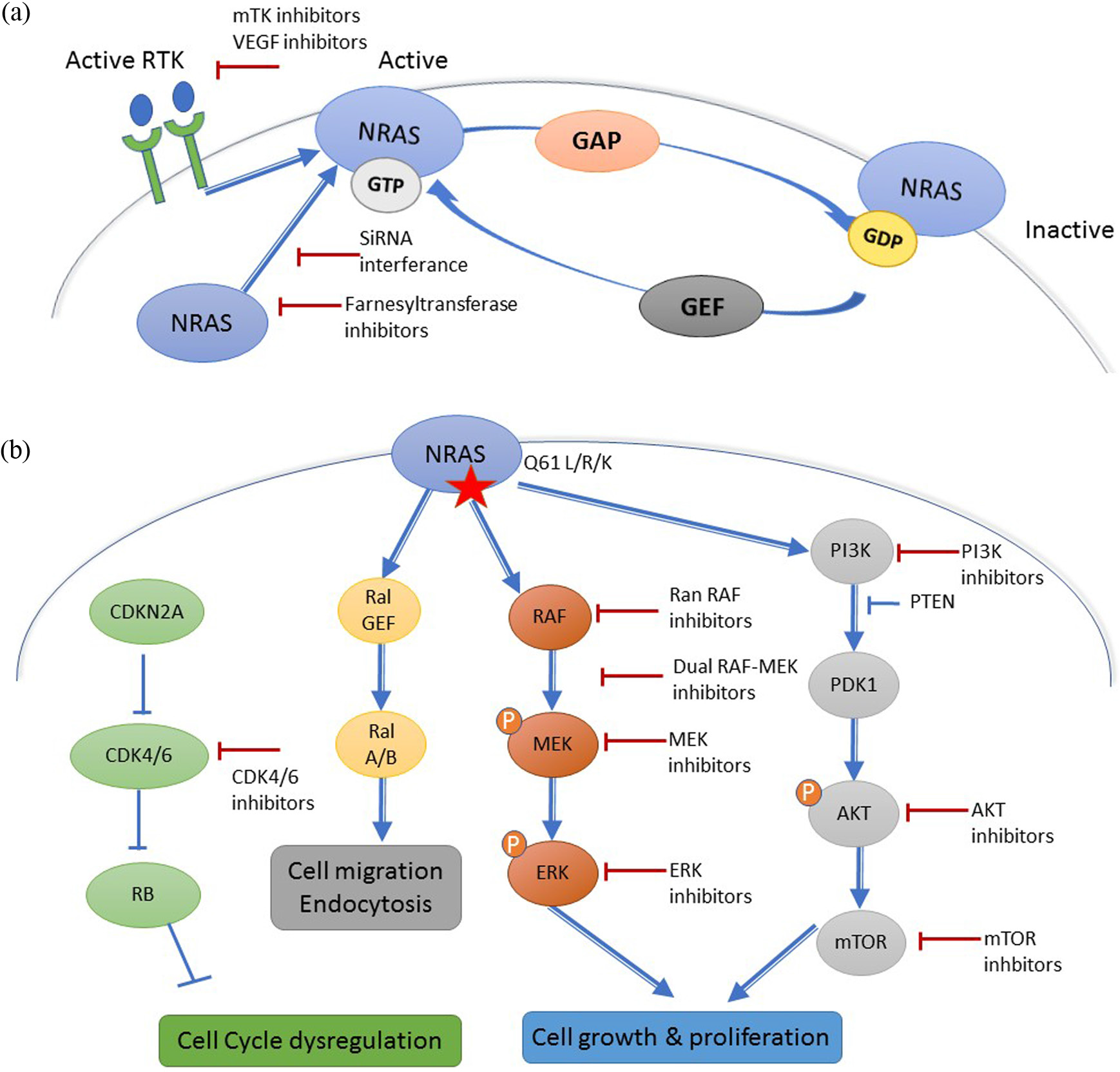
(a) Mechanism of NRAS activation. Receptor tyrosine kinase (RTK)-mediated activation requires dissociation of protein-bound GDP, a process that is accelerated by guanine nucleotide exchange factors (GEFs). The hydrolysis of GTP to GDP, that inactivates NRAS is accelerated by GTPase activating proteins (GAPs). (b) Downstream effectors of NRAS and different targeted therapy strategies.
Activated RTKs stimulate the passage from the inactive RAS-GDP to the active RAS-GTP with the help of guanine nucleotide exchange factors (GEFs), such as Son of Sevenless Ras/Rho Guanine Nucleotide Exchange Factor (SOS) that catalyze the exchange of Guanosine diphosphate (GDP) for GTP. 7 GTPase activating proteins (RAS-GAPs), such as neurofibromin (NF1), inactivate RAS-GDP, and are considered as tumor suppressors. 7 Activated RAS proteins stimulate different cell signaling pathways like the MAPK signaling pathway, the phosphoinositide 3-kinase (PI3K)/AKT pathway, and other factors like the RAL guanine nucleotide exchange factors (RAL-GEFs) 8 (Figure 1a).
NRAS is very rarely mutated in uveal melanoma. 9 In cutaneous melanoma, NRAS is most frequently mutated at hotspots in exon 1 (codon 12) and exon 2 (codon 61) which results in the prolongation of its active GTP-bound state. 10 A glutamine to arginine/lysine/leucine substitution at position 61 (Q61R/K/L) accounts for 80% of all NRAS mutations in melanoma. 9 No distinct clinical behavior was identified between NRAS exon 1 and exon 2 mutations. 11 A BRAF V600E and an activating NRAS mutation were generally believed to be mutually exclusive, but can rarely occur in less than 1% of treatment-naïve melanoma patients. 9
Contrarily to BRAF that is frequently mutated in benign nevi, NRAS is rarely mutated in benign melanocytic lesions, except in congenital nevi. 12 At the time of initial diagnosis, NRAS-mutant cutaneous melanomas are generally located on the extremities, in older patients with more markers of chronic sun damage than BRAF-mutant melanoma 13 even though the prevalence in older patients is disputed. 14 Histologically NRAS-mutated melanomas are more frequently associated with a nodular subtype than BRAF melanomas, which are more frequently associated with an Superficial Spreading Melanoma (SSM) subtype. 13 In patients with a metastatic disease, NRAS and BRAF mutations are associated with a higher risk of central nervous system involvement compared with WT BRAF and NRAS melanoma. 9 Generally NRAS mutations are associated independently with decreased overall survival compared with WT melanoma 9 even though these results have not been confirmed in all studies.8,11
Directly targeting NRAS
Due to the extremely high affinity of RAS to GTP and GDP, and the high intracellular concentrations of GTP, developing drugs that effectively compete with the nucleotide generally is considered to be an unrealistic approach. 15 Most attempts to directly target RAS have focused on inhibiting the hydrolysis of GTP to GDP by trying to identify antagonists of GEFs or drug-like mimics of RAS-GAPs 16 (Figure 1a). Until now these efforts have been largely unsuccessful, but research of a direct RAS-targeted therapy is still very active and recently small compounds that bind directly to the G-domain with inhibitory effects on mutated RAS function have been discovered and might permit the development of such drugs in the future. 17
To be active, NRAS has to undergo post-translational modifications, like the farnesylation of a cysteine residue that permits its insertion to the plasma cell membrane where it is activated. 18 Initial in vivo data suggested that farnesyl transferase inhibitors (FTIs) could reduce tumor growth in RAS-driven breast cancer and lymphoid tumors 19 and that the FTI lonafarnib, could sensitize melanoma cells to RTK inhibitors like sorafenib. 20 Unfortunately, these results were not confirmed in the clinical setting where two FTIs, lonafarnib and tipifarnib, progressed to advanced clinical trials but failed to show efficacy against NRAS and KRAS-driven cancers.16,21,22 Farnesyl transferase inhibition is considered to have failed in the clinics because, in the presence of FTI, NRAS and KRAS become substrates for geranylgeranyltransferase I (GGTase I) through a process known as alternative prenylation, and FTIs therefore do not effectively block RAS attachment to the plasma membrane. 23 Dual FTI and GGTase I inhibitors have been tested in the clinical setting, but their development is limited by their toxicity. 24 Other approaches to inhibit the localization of RAS to the plasma membrane have been attempted or are currently being evaluated in the preclinical or clinical setting but most of them are limited by toxicity 16 or technological issues such as how to deliver siRNA using nanoparticle-based delivery systems. 25
Targeting upstream effectors of NRAS
Under physiologic conditions, the interaction between an RTK and its ligand induces RTK dimerization, trans-phosphorylation, and activation which in turn stimulates RAS by recruiting GEFs (Figure 1a).
Tyrosine kinase inhibitors (TKIs) and monoclonal antibodies targeting upstream regulators of RAS have been tested in melanoma with limited clinical benefits when used as single agents (Table 1). Targeting downstream NRAS effectors has been associated with an upregulation of RTKs like EGFR, HER3, and ERRB3 in NRAS-mutated melanoma. 26 Targeting RTKs with a TKI might therefore be efficacious in combination with MAPKi or PI3K-AKT-mTOR inhibitors to avoid targeted therapy-acquired resistance and avoid compensatory reactivation via RTK signaling. 27
Ongoing and completed clinical trials testing mTKI in melanoma and advanced solid tumors.

Red trials considered negative by the authors.
Green trials considered positive by the authors.
No published article.
(…) corresponds to the reference of the article that published the results of the study.
For instance sorafenib, a multi-TKI (mTKI), showed no clinical activity in melanoma when tested as a single agent 28 or in combination with chemotherapy (C/P, DTIC, TMZ29–31), but combinations of sorafenib with Mesenchymal epithelial transition factor receptor (MET) inhibitors or with alpha-mangostin might be more promising to treat NRAS-mutant melanoma and are currently being tested in early clinical trials 32 and in preclinical experiments. 33
Axitinib and pazopanib, two other mTKIs, showed more promising results in phase II clinical trials in BRAF WT melanoma both when used as single agents 34 and when used in combination with C/P,35,36 but have shown no benefit in NRAS-mutated melanoma. Ongoing trials are evaluating the safety of combining pazopanib with the MEKi trametinib [ClinicalTrials.gov identifier: NCT01438554].
The mTKI lenvatinib is currently being tested as a single agent in melanoma [ClinicalTrials.gov identifier: NCT01136936]. When lenvatinib is combined with TMZ, it has no clinical benefit, 37 however, combined with DTIC it shows promising results in a phase II trial on metastatic melanoma, but not specifically in NRAS-mutant melanoma [ClinicalTrials.gov identifier: NCT01133977]. It is also currently being tested in combination with cMET inhibitor E7050 [ClinicalTrials.gov identifier: NCT01433991] and in combination with anti-PD1 pembrolizumab [ClinicalTrials.gov identifier: NCT02501096] in two phase I–II trials. Amuvatinib shows promising preclinical data in NRAS-mutant melanoma. 38
Bevacizumab, a monoclonal antibody against VEGF-A was showed to be safe in phase II studies where it was combined with DTIC 39 C/P,40,41 TMZ, 42 fotemustine, 43 everolimus, 44 temsirolimus, 45 ipilimumab, 46 erlotinib 47 or imatinib 48 with limited clinical activity in NRAS-mutant melanoma. Vatalanib, another VEGF antibody seems to have no clinical activity as a single agent 49 and is currently being tested in combination with everolimus [ClinicalTrials.gov identifier: NCT00655655].
Anti-integrin alphavbeta antibodies (etaracizumab, intetumumab) showed limited clinical activity compared with DTIC in metastatic melanoma.50,51
Targeting downstream effectors of NRAS
Targeting the MAPK signaling pathway with single agents
RAS activates the MAPK signaling pathway by inducing a conformational change and activation of BRAF, CRAF or ARAF. 52 Upon activation, homo or heterodimers of RAF phosphorylate MEK that then phosphorylates the transcription factor ERK that enters the nucleus and activates cell behaviors like proliferation and differentiation 53 (Figure 1b). Targeting RAS MAPK downstream effectors therefore seems a promising approach.
BRAFi were the first targeted therapies to be approved for BRAF-mutant melanoma. 6 Unfortunately, first and second generation BRAFi cannot be used as single agents to treat NRAS-mutant melanoma because while these inhibitors are effective at shutting down ERK signaling mediated by mutant-BRAF, they paradoxically upregulate ERK activity in the presence of oncogenic RAS, by stimulating BRAF–CRAF heterodimerization. 53 Pan RAF inhibitors (PRis) (TAK-632, LY3009120, Compound A) show interesting preclinical data in this setting, 54 as single agents 55 but also in combination with MEKis. 56 Ongoing phase I clinical trials are testing PRi alone (CCT3833 in ClinicalTrials.gov identifier: NCT02437227; LY3009120 in ClinicalTrials.gov identifier: NCT02014116) or in combination with an anti-PD1 antibody (LXH254+PDR001, ClinicalTrials.gov identifier: NCT02607813).
RAF can also be inhibited in NRAS-mutant melanoma with a dual RAF/MEKi (RO5126766) that stabilizes the RAF-MEK dimer and therefore blocks the phosphorylation and release of RAF. 57 A phase I study showed that RO5126766 has manageable toxicity with encouraging preliminary antitumor activity. 58
Targeting MEK1/2 in NRAS-mutated melanoma is currently the most developed targeted therapy approach. MEKis are orally bioavailable and either ATP-competitive or non-ATP-competitive, allosteric binding inhibitors of MEK. 59
The first generation of MEKis (CI-1040, PD-901) showed limited clinical benefit in unselected melanoma patients as single agents60,61 and also in combination with docetaxel in WT melanoma. 62 Second and third generation MEKis (trametinib, binimetinib, selumetinib, pimasertib, cobimetinib) seem to have a safer toxicity profile and a more promising clinical activity and are therefore being tested in phase II/III clinical trials. 59
In a phase III clinical trial, the MEKi binimetinib recently showed its superiority compared with DTIC in NRAS-mutant melanoma (SMR, 2016; 63 ASCO, 2016 64 ) even though its benefit was small [progression-free survival of 2.8 months with binimetinib compared with 1.5 months for DTIC; hazard ratio (HR) 0.62 (95% confidence interval (CI) 0.47–0.8)]. Pimaseritib showed promising results in a phase I clinical trial and is being in compared with DTIC in NRAS-mutated melanoma, in a completed but not published phase II trial [ClinicalTrials.gov identifier: NCT 0193068]. Trametinib is United States Food and Drug Administration (US FDA)-approved in combination with dabrafenib in BRAF-mutant melanoma. It has not been specifically tested in NRAS melanoma, but in a phase I trial for unselected melanoma patients, two out of seven patients with an NRAS-mutated melanoma achieved stable disease with trametinib treatment. 65
According to preclinical data, the next generation MEKi GDC-0623 [ClinicalTrials.gov identifier: NCT01106599] and G-573 might be more effective in RAS-mutated cells compared with BRAF-mutated cells 66 but TAK-733 showed limited tumor activity in a phase I trial, with no further development currently planned. 67
Finally, preclinical data suggest that ERK inhibitors (ERKis) might be interesting in NRAS-mutant melanoma as ERK represents the final single node in the MAPK signaling pathway for potential inhibition. 68 Several ERKis are currently being developed in the preclinical setting and in phase I clinical trials as single agents or in combination with chemotherapy or MEKis: BVD-523 [ClinicalTrials.gov identifiers: NCT02608229, NCT02296242], SCH772984, 69 LTT462 [ClinicalTrials.gov identifiers: NCT02711345]; CC-90003 [ClinicalTrials.gov identifier: NCT0231012] and GDC 0994 [ClinicalTrials.gov identifiers: NCT01875705, NCT02457793]. 70
Combining MAPKi and the PI3K-AKT-mTOR inhibitors
NRAS not only activates the MAPK signaling pathway, but also activates the PI3K-AKT-mTOR cell signaling pathway, RAL pathways and cell cycle regulatory proteins. 8 This may explain why MEKis as single agents are less effective in NRAS-mutated melanoma than BRAFis are in BRAF-mutant melanoma. 8
Multiple classes of inhibitors of PI3K-AKT-mTOR are available including PI3K inhibitors (pan-isoform and isoform specific), dual PI3K-mTOR inhibitors, AKT inhibitors (AKTis) and mTOR inhibitors (mTORC1 and mTORC1/2) (mTORis). 71 Combining MAPKis with these inhibitors demonstrated promising preclinical in vitro and in vivo results in NRAS-mutant melanoma, 72 however, these results have yet to be translated into the clinical setting. Many clinical trials combining MAPKis and inhibitors of PI3K-AKT-mTOR have been or are currently being tested (Table 2). Unfortunately, this approach is limited by overlapping toxicities and compensatory signaling within and between cell signaling pathways that results in insufficient plasma drug levels of PI3-AKT-mTOR inhibitors for antitumor activity.73–75 This could be overcome by intermittent high dose administration of PI3K-AKT-mTOR inhibitors associated with continuous MEKi administration as suggested by preclinical data. 76
PI3K-AKT-mTOR inhibitors that have been tested in combination with MEK inhibitors in the clinical setting.

AZD2014 (vistusertib); AZD6244 (selumetinib); BAY80-6946 (copanlisib); BAY86-9766 (refametinib); BEZ235 (dactolisib); BKM120 (buparlisib); BYL719 (alpelisib); CCI-779 (temsirolimus); GDC-0068 (ipatasertib); GDC-0941 (taselisib); GDC-0973 (cobimetinib); GSK1120212 (trametinib); GSK2110183 (afuresertib); GSK2141795 (uprosertib); GSK2126458 (omipalisib); MEK162 (binimetinib); MSC1936369B (pimasertib); RAD001 (everolimus); SAR245409 (voxtalisib).
Combining MAPKis with cell cycle regulator protein inhibitors
NRAS induces the expression of cyclin D1 that regulates cell cycle regulators like cyclin-dependent kinase 4/6 (CDK4/6) 77 that are fundamental to RAS-induced transformation. 8 CDK4/6 inhibitors are currently being tested in combination with MEKis with encouraging early clinical results. Ribociclib (LEE011) is being tested in combination with binimetinib in an encouraging phase II trial 78 and palbociclib is being tested in combination with trametinib. 79
Wee1 is a kinase that inactivates the Cyclin B Cell division control protein kinase (CDC)/cyclin B complex that regulates the G2 cell cycle checkpoint. 80 Combining Wee1 inhibitor with an mTOR inhibitor like rapamycin has shown promising preclinical data in NRAS-mutated melanoma 81 and the Wee1 inhibitor AZD-1775 is currently being tested in phase I trials [ClinicalTrials.gov identifiers: NCT02610075; NCT02617277].
Polo-kinase 1 (PK1) is overexpressed in NRAS-mutant melanoma and regulates the cell cycle. 82 PK1 inhibitors have shown disappointing clinical activity as single agents, but preclinical data suggest they may be interesting in combination with MEKi in NRAS-mutant melanoma.83,84
Combining MAPKis and RalGEF inhibitors
RAS activates the RalGEF pathway. TANK-binding kinase 1 (TBK1)is activated downstream of RALB and has shown promising preclinical activity in NRAS-mutant melanoma when combined with MEKis.85,86
Other combination of targeted therapies
ROCK 1/2 are RHO GTPase-activated serine/threonine kinases that are involved in RAS tumor proliferation. Preclinical data suggest that ROCK inhibition could increase MEKi antitumoral activity in vivo. 87
Preclinical data suggests combining ERβ inhibition with MAPKi or PI3K-AKT-mTOR inhibition could be interesting in NRAS melanoma. 88
Targeting the immune system
The arrival of immune-based therapies for the treatment of melanoma has revolutionized the standard of care and are now the first-line treatment for NRAS and WT melanoma. 89 Interleukin (IL)2 and anti-CTLA4 antibody (ipilimumab) were the first immunotherapies approved by the US FDA to treat metastatic melanoma, with durable responses seen in 5–15% of patients despite severe acute toxicities.90,91 More recently therapeutic approaches aimed at activating antitumor immunity through blockade of the immune checkpoint PD1 with nivolumab and pembrolizumab have showed objective responses in 25–50% of patients in early trials.92,93
Due to a distinct immune microenvironment compared with BRAF-mutant melanoma, 94 NRAS-mutant melanoma may be associated with more frequent responses in patients treated by IL2, ipilimumab, and anti-PD1.95–97
In vitro and in vivo, MEKis enhance melanoma antigen expression and reactivity to antigen-specific T-lymphocytes leading to a synergy with immune checkpoint blockade in murine models.98,99 This gives a strong rationale to combine targeted and immune-based strategies 100 in NRAS-mutated melanoma with numerous ongoing trials. 101
Conclusion
NRAS has often been considered an undruggable target because even though its role in cancer has been demonstrated for more than 25 years, no targeted therapy has been approved despite extensive efforts in melanoma and other RAS-mutated malignancies. This has recently changed with the advent of new MEKis that are tested in combination with a variety of drugs that use different approaches: inhibition of upstream RAS effectors, inhibition of PI3K-AKT-mTOR, inhibition of cell cycle regulators and activation of anti-tumor immunity. As each combination of treatments pursues its clinical development though phase I, II and III clinical trials, the challenge will be to choose in what order to use them in NRAS-mutant melanoma patients.
Footnotes
Funding
Amélie Boespflug is currently being funded by a graduate grant of the Fondation pour la Recherche Médical (FRM), France (FDM20150633361).
Conflict of interest statement
The authors declare that there is no conflict of interest.