
Abstract
DNA repair genomic aberrations in the Homologous Recombination pathway are identifiable in up to 25% of patients with advanced prostate cancer, making them more likely to benefit from treatment with poly (ADP-ribose) polymerase inhibitors (PARPi) alone or in combination with other therapies, particularly when BRCA driver genomic aberrations are documented. Although several clinical trials have demonstrated the efficacy of this approach, the validation of reliable biomarkers predictive of response still needs further improvement to refine patient selection. In this setting, the characterization of resistance mechanisms and the validation of novel biomarkers are critical to maximize clinical benefit and to develop novel treatment combinations to improve outcomes. In this review, we summarize the development of PARPi in prostate cancer as single agent as well as the efficacy of their combination with other drugs, and the future directions for their implementation in the management of advanced prostate cancer.
Plain language summary
Prostate cancer is the most common cancer in men worldwide. Alterations in the genes responsible for repairing damaged DNA are found in up to 25% of advanced prostate cancer patients. This inability of cells to repair damaged DNA allows tumours to grow, but it is also exploited by new treatments. An example of such therapies are the inhibitors of the Poly-ADP ribose polymerase, known as PARP inhibitors. PARP inhibitors are being developed alone and in combination with other drugs for the treatment of prostate cancer. In this manuscript, we provide an overview of the studies conducted in prostate cancer, as well as the future directions of PARP inhibitors for the management of the disease.
Introduction
In men, prostate cancer is the most frequently diagnosed cancer type and the second cause of cancer death worldwide. 1
The disease is predominantly androgen-dependent, and even most advanced prostate cancers initially respond to androgen receptor (AR) blockage. However, metastatic prostate cancer is a lethal, molecularly heterogeneous disease characterized by its lack of durable responses in the advanced setting. For that reason, prostate cancer molecular characterization has been crucial to understand the adaptive responses to anti-androgen therapy, chemotherapy, targeted therapies, and immunotherapy, leading to the development of new therapeutic strategies for these patients. 2 In this setting, the DNA damage repair (DDR) pathway has proven to be an attractive target, with several clinical trials demonstrating the efficacy of its modulation in terms of biochemical, tumoral and radiological response, together with overall survival (OS).3–8
In this review, we summarize the pivotal trials which have, so far, tested the efficacy of modulating this pathway, particularly the Homologous Recombination pathway in metastatic castration-resistant prostate cancer (mCRPC) patients. We also give insight into the potential limitations of these approaches when being implemented in daily clinical practice. Finally, we propose some directions to improve patient selection and clinical outcomes in future.
DDR pathway in prostate cancer
The DDR pathway is responsible for maintaining genomic stability when cells are exposed to DNA-damaging agents. 9 In these situations, DDR proteins target DNA lesions by modulating transcription and transduction signals, cell-cycle checkpoints, and other cellular processes. 9
DNA repair systems can be divided into the following major entities 10 : (1) base excision repair, (2) nucleotide excision repair, (3) mismatch repair, (4) recombinational repair, which is further divided into homologous recombination repair (HRR) and non-homologous end joining (NHEJ). HRR is a very complex, high-fidelity pathway that restores the original DNA code in an error-free mode but requires a sister chromatid as a template, thus it is restricted to the S and G2 phases of the cell cycle. Key mediators of HRR include BRCA1 and BRCA2 not only are key mediators in the HRR pathway but are also involved in the repair of DNA double-strand breaks mediated by other pathways, such as Fanconi Anemia and NHEJ. 9 NHEJ is error-prone and might introduce new mutations. Balance between HRR and NHEJ is key for the maintenance of genomic stability.
DNA repair aberrations that arise during tumor development can make some cancer cells reliant on some of the above-mentioned pathways for survival. 9 In addition, cancer treatments induce cell death by causing direct or indirect DNA damage. 11 In these situations, DNA damage response proteins activate inter-related molecular signals to recognize DNA damage, mediate DNA repair and maintain the integrity of the genome. 9 Considering that alterations in genes involved in DDR are identifiable in at least 25% of prostate cancer patients, multiple efforts are being conducted to target this vulnerability of tumors.
Poly (ADP-ribose) polymerase 1 and 2 (PARP1 and PARP2) enzymes are key to DDR, acting as DNA damage sensors and signal transducers to repair DNA lesions and initiating repair of DNA single-strand breaks (SSBs). 12 PARP inhibitors (PARPi) have two general effects: catalytic inhibition of PARP and trapping PARP on damaged DNA, therefore preventing the access of other DNA repair proteins. PARPi shows variable ability to inhibit the catalytic activity and to trap PARP, resulting in different antitumor activity. 10
PARP inhibition enables the accumulation of SSBs which can progress to double-stranded breaks, usually repaired through HRR (Figure 1). 13 PARPi, therefore, induces cancer cell death through “synthetic lethality,” where the combination of PARPi and HRR deficiencies (like BRCA pathogenic genomic aberrations) is lethal due to unrepairable DNA damage (Figure 1).12,13

PARPi mediated synthetic lethality.
Prevalence and clinical implications of DDR pathogenic genomic aberrations and PARPi in prostate cancer
Deleterious genomic aberrations in DDR genes have been described in 10% of localized prostate tumors and up to 30% of mCRPC patients.5,13,14 Among the DDR pathways, the HRR, is the one most frequently impaired in advanced prostate cancer. In the PROfound study, 28% of the samples successfully analyzed had at least one HRR alteration. The most frequently altered gene was BRCA2 (8.7%), followed by CDK12 (6.3%), ATM (5.9%), CHEK2 (1.2%), and BRCA1 (1%). In 2.2% of cases, aberrations in two or more genes were detected. 5 However, HRR alterations prevalence differs across studies. A recent analysis 15 of over 14,000 prostate tumors identified HRR alterations in 14%, of which BRCA mutations were the most common (5.4%), followed by alterations in ATM, CHEK2, and CDK12. Although a similar frequency of HRR alterations was noted across populations with different genetic backgrounds, some gene prevalence (i.e., CDK12) varied.
Previous studies analyzing paired primary and metastatic samples obtained at the time of castration resistance have suggested that these events may occur early in the course of the disease.16–18 As we will discuss below, this raises the question of whether treatment with PARPi at earlier stages of prostate cancer could improve these patients’ outcomes. In this setting, germline and/or somatic screening for these aberrations at diagnosis would be key to improve patient selection and to advance to a more personalized, targeted-driven therapy.
In the Capture study, pathogenic genomic aberrations in HRR genes, particularly in BRCA, were associated with worse outcomes in terms of progression-free survival (PFS) and OS in mCRPC patients treated with either an androgen receptor signaling inhibitor (ARSi) or taxanes in the first-line setting, compared to non-HRR and HRR non-BRCA-mutated patients. In an exploratory analysis, these results seemed to be independent of germline versus somatic, bi-allelic versus mono-allelic, or BRCA2 versus BRCA1 genomic aberrations. 19
PARPi in monotherapy in mCRPC
As discussed above, deleterious HRR alterations are frequent in mCRPC, and early clinical trials detected significant antitumor responses in some molecularly selected patients. This paved the way for the development of PARPi in this scenario. The pivotal trials testing the efficacy of PARPi monotherapy in mCRPC are summarized below and in Table 1.
Summary of phase II trials with PARPi in mCRPC.

ARSi, androgen receptor signaling inhibitors; CTC, circulating tumor cell; DDR, DNA damage repair; mCRPC, metastatic castration-resistant prostate cancer; ORR, objective response rate; PARPi, poly (ADP-ribose) polymerase inhibitors; PSA, prostate-specific antigen.
TOPARP-A was a discovery phase II trial designed to identify biomarkers predictive of response to the PARPi olaparib in mCRPC. 3 This study enrolled 50 molecularly unselected, mCRPC patients progressive to standard treatments for their metastatic disease. Primary endpoint was composite response rate (CRR), defined either as objective response rate (ORR) by response evaluation criteria in solid tumors 1.1 (RECIST 1.1), a decline in prostate-specific antigen (PSA) level of at least 50% (PSA50), and/or conversion in circulating tumor cell count from ⩾5 cells/7.5 mL to < 5 cells/7.5 mL of blood. Patients were considered to be biomarker-positive when a homozygous deletion or deleterious mutation was identified in any of the DNA repair genes analyzed. Overall, 32% of evaluable patients fulfilled the prespecified definition of response, and almost all responders were biomarker positive, including seven patients harboring somatic or germline BRCA2 alterations. Based on these results, the second stage of the trial (TOPARP-B) 20 was designed to include only those patients with the DDR defects previously associated with PARPi sensitivity in TOPARP-A. All subsequent studies with PARPi in monotherapy were limited to patients with DDR alterations.
The TOPARP-B trial randomized 98 mCRPC patients with DDR pathogenic genomic aberrations to two different doses of olaparib (400 mg vs 300 mg twice a day). 20 The overall CRR was 54.3% in the 400 mg cohort versus 39.1% in the 300 mg cohort. The best responses were observed in the BRCA1/2 subgroup, with a CRR of 83.3%, an ORR of 52.4% and a median PFS of 8.3 months.
Based on these results, the TRITON2,4,21,22 TALAPRO-1 7 and GALAHAD 6 phase II trials tested the antitumor activity of other PARPi in heavily pre-treated mCRPC patients with HRR pathogenic genomic aberrations. Overall, a significant benefit was seen in the BRCA 1/2 population, with the benefit for other genomic aberrations being less consistent.
The TRITON phase II trial tested the antitumor activity of the PARPi rucaparib in patients with mCRPC and deleterious germline or somatic alterations in patients with DDR alterations, previously treated with ARSi and taxane-containing therapies. In patients with BRCA1/2 alterations, treatment with rucaparib 600 mg twice daily showed promising results in terms of ORR (43.5%) and PFS (9 months). 4
Similarly, the phase II TALAPRO-1 trial recruited mCRPC patients with genomic aberrations in HRR-related genes whose disease had progressed after one or two taxane-based chemotherapies and enzalutamide, abiraterone, or both. 7 Eligible patients were given oral talazoparib 1 mg per day until disease progression or unacceptable toxicity. The primary endpoint was confirmed ORR, defined as the best overall soft-tissue response of complete or partial response per RECIST 1.1, by blinded independent central review. Confirmed ORR was 29.8%, and confirmed ORR by hierarchical stratification of the HRR-deficient group was seen in 46% of patients with BRCA2 alterations (n = 57), 50% of BRCA1 alterations (n = 4), 25% of PALB2 alterations (n = 4), and 12% of ATM alterations (n = 17). 8
Finally, the GALAHAD trial tested the antitumor activity of niraparib in mCRPC patients with progression on a previous ARSi and a taxane, and biallelic alterations in DDR genes, including BRCA1/2, ATM, FANCA, PALB2, BRIP1, and HDAC2, assessed in blood, tumor tissue, or saliva. The primary endpoint of ORR in the measurable BRCA cohort was 34.2%, with median duration of response of 5.55 months. 6
These promising results supported the design of the phase III trials PROfound and TRITON3.
PROfound5,23 was the first randomized phase III biomarker-driven trial to assess the efficacy of the PARPiolaparib compared to a second ARSi in mCRPC patients with deleterious genomic alterations in the DDR-HHR pathway and disease progression after receiving a prior ARSi in the castration-sensitive or resistant setting. The primary endpoint was radiologic progression-free survival (rPFS) in Cohort A and in the overall population. Depending on their genomic aberrations, patients were allocated to Cohort A (BRCA1, BRCA2, or ATM alterations) or Cohort B (other genes assessed). Olaparib improved rPFS with an absolute increased survival of 3.8 months in cohort A and 2.3 months in the overall population. Olaparib also improved OS in Cohort A, but not in Cohort B. Although 66% of patients were crossed over to the experimental arm at disease progression, a sensitivity analysis adjusted for this crossover kept showing a 58% decrease in the risk of death for patients in Cohort A. A gene-by-gene exploratory analysis suggested that better clinical outcomes were driven by the BRCA2 population, with less clear benefit in other aberrations. After these results, the FDA approved olaparib for patients with alterations in the HRR genes tested in the PROfound study, whereas EMA restricted the approval for patients with BRCA1 and BRCA2 genomic aberrations.
The TRITON-3 study assessed the efficacy of rucaparib monotherapy in mCRPC patients with BRCA1, BRCA2, or ATM pathogenic genomic aberrations after progression to an ARSi. 8 Patients were randomized to receive rucaparib or physician’s choice of treatment (docetaxel or a second ARSi). The primary outcome was median duration of imaging-based PFS according to an independent review. Median rPFS in the intention to treat population was significantly longer in the rucaparib than in the control arm. Similarly to PROfound, differences in rPFS were driven by the BRCA population, without significant benefits in ATM mutated patients. When comparing rucaparib and docetaxel, patients with BRCA1/2 alterations had longer rPFS, without significant differences in ATM patients. Similarly to the PROfound study, rucaparib resulted in improved rPFS in the BRCA population when compared to a second ARSi, with the benefit in ATM patients being less consistent. Mature OS are still pending. Based on these results, FDA has approved rucaparib for the treatment of BRCA-mutated mCRPC after progression on ARSi.
PARPi in combination with ARSi
According to previous studies, AR can transcriptionally stimulate the expression of DDR genes, 24 while PARP1 promotes AR transcriptional function,25,26 leading to the hypothesis that concomitant inhibition of AR and PARP would result in increased antitumor activity. Li et al. reported that enzalutamide reduced the expression of a variety of HRR genes in AR-dependent cell lines and so did olaparib in both AR-dependent and AR-independent cell lines. 24 In fact, treatment with enzalutamide followed by olaparib increased antitumoral activity in murine prostate cancer xenografts compared to monotherapy with either agent. Therefore, PARP inhibition might downregulate AR signaling and promote a longer hormone-sensitive disease status. This molecular rationale has paved the way for the development of phase III trials testing the efficacy of different combinations of PARPi and ARSi. So far, three randomized phase III trials (PROPEL,27,28 TALAPRO-2,29,30 and MAGNITUDE31,32) have assessed the efficacy of this approach as first-line treatment of mCRPC
In the PROpel trial27,28 796 mCRPC patients were randomized to receive olaparib and abiraterone acetate plus prednisone versus placebo and abiraterone as first-line treatment for mCRPC. Prior docetaxel in the metastatic hormone-sensitive (mHSPC) setting was allowed, but patients had to be ARSi naïve. Stratication analysis included site of distant metastases and prior treatment with docetaxel. HRR status was retrospectively analyzed in tumor and plasma samples. Patients were classified as HRR mutant (28.4%), non-HRR mutant (69.3%) or unknown (2.3%), with their baseline characteristics being balanced between treatment arms. Although a benefit in rPFS was observed in the overall population with olaparib plus abiraterone (24.8 months vs 16.6 months; HR 0.66, 95% CI 0.54–0.81) this benefit was again driven by BRCA-altered patients (HR 0.23, 95% CI 0.12–0.43) followed by the HRR mutated (HRRm) population (HR 0.50, 95% CI 0.34–0.73), and the non-HRRm subgroup (HR 0.76; 95% CI 0.60–0.97). An OS benefit was only observed in the HRRm population, (HR 0.66, 95% CI 0.66–0.95) and particularly in those with BRCA alterations (HR 0.29; 95% CI 0.14–0.56). In fact, the 7.4 months improvement in OS in the overall population (HR 0.81, 95% CI 0.67–1) was not statistically significant. Most frequent adverse events (AEs) in the combination group included anemia (46%), fatigue (37.2%), and nausea (28%), and most frequent grade ⩾3 AEs were anemia (15%), venous embolisms (6.8%), hypertension (3.5%), and fatigue (2.3%). Based on these results, olaparib plus abiraterone was approved by EMA as first-line therapy for mCRPC patients regardless of HRR status, while FDA restricted the approval to BRCA1 and BRCA2 mCRPC patients.
The TALAPRO-2 trial29,30 assessed the efficacy of the combination of enzalutamide plus talazoparib as first-line therapy for mCRPC. Patients were stratified by prior treatment with docetaxel or abiraterone in the metastatic-sensitive setting and by HRR status. In Cohort 1, 805 patients were prospectively tested for HRR status on tumor tissue samples. Median rPFS was significantly longer for the overall population in the experimental arm, with a higher benefit in the HRRm population (HR 0.46, 95% CI 0.3–0.7) (Table 2). Exploratory analysis in patients without HRR aberrations also showed a benefit in rPFS in the experimental arm, further supporting a potential synergy from the concomitant inhibition of AR and PARP (Table 2). Cohort 2 in TALAPRO-2 (n = 230) expanded the HRR-deficient population to 399 patients, of whom 155 had BRCA1/2 alterations. In this population, the rPFS benefit from the combination was confirmed (HR 0.44, 95% CI 0.32–0.60). Similarly to PROPEL, in TALAPRO-2 the greatest benefit in terms of clinical outcomes was also driven by the BRCA patients, with the benefit in other aberrations being more modest (Table 2). Anemia (65%) was the most frequent side effect reported in the experimental arm, followed by neutropenia (36%), fatigue (34%), thrombocytopenia (25%), hyporexia (22%), and nausea (21%). Grade ⩾3 hematological toxicities were frequent with the combination (anemia 46%, neutropenia 18%, thrombocytopenia 7%). Talazoparib plus enzalutamide is currently approved by FDA and as first-line therapy for HRR-deficient mCRPC patients, while EMA has approved it for all mCRPC patients.
Design, characteristics, and outcomes of MAGNITUDE, PROPEL, and TALAPRO-2 phase III studies.

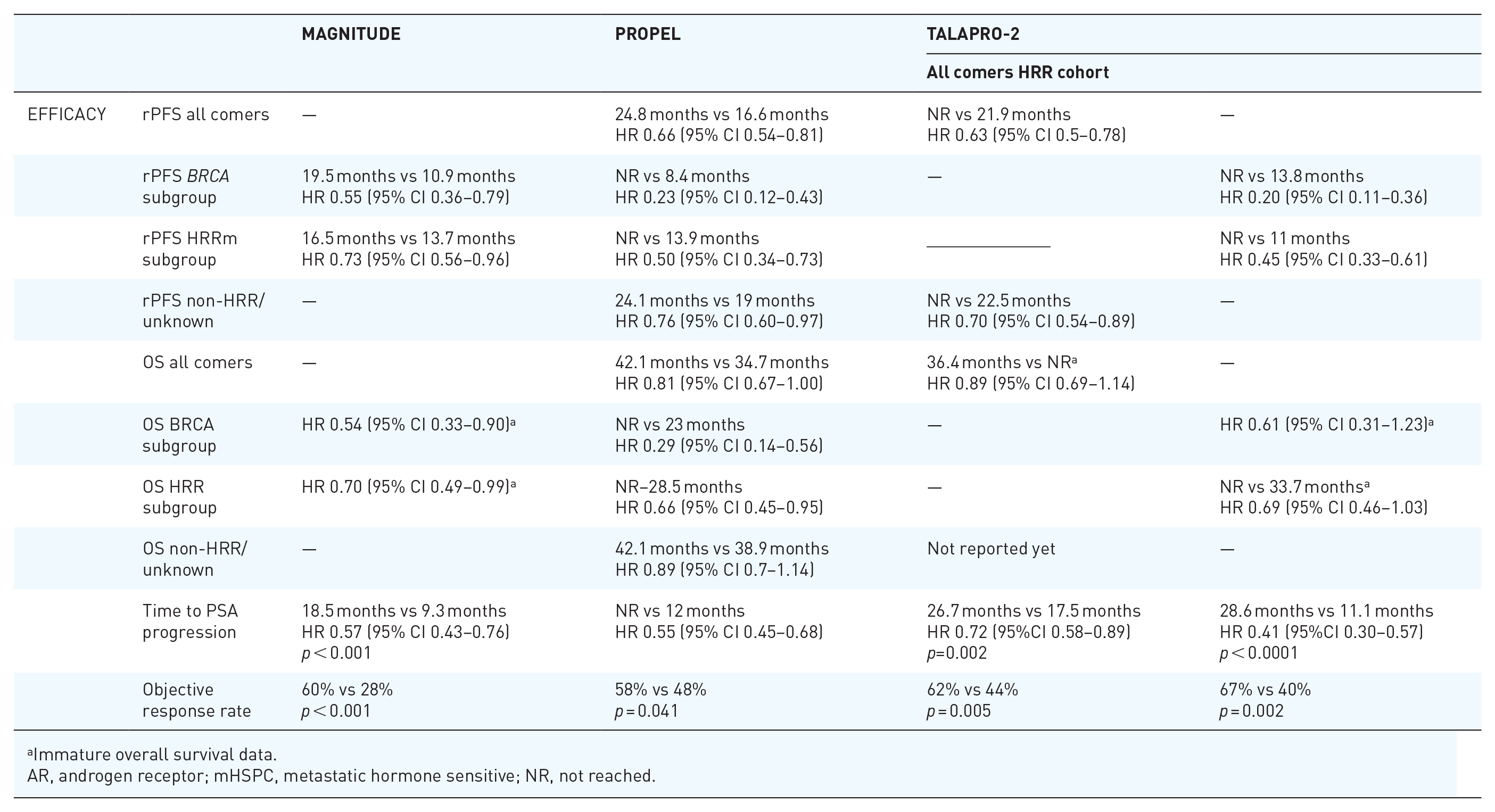
Immature overall survival data.
AR, androgen receptor; mHSPC, metastatic hormone sensitive; NR, not reached.
The MAGNITUDE trial31,32 tested the efficacy of combining niraparib plus abiraterone as first-line treatment for mCRPC patients. A prospective screening for ATM, BRCA1, BRCA2, BRP1, CDK12, CHEK2, FANCA, HDAC2, and PALB2 deleterious aberrations was conducted. Biomaker-positive patients (those with HRR alterations) were included in Cohort 1 while Cohort 2 included biomarker-negative patients. Abiraterone prior to randomization was allowed for up to 4 months while the molecular screening was ongoing. Stratified analysis included run-in treatment with abiraterone and prior mHSPC treatment. A pre-planned futility analysis suggested a lack of benefit from the combination in Cohort 2, which therefore was halted. Of the 423 patients in Cohort 1, 190 (45%) had BRCA1/2 alterations. Niraparib plus abiraterone increased rPFS (16.5 months vs 13.7 months; HR 0.76, 95% CI 0.6–0.97) in HRR-altered patients, particularly in the BRCA1/2 subgroup (16.6 months vs 10.9 months; HR 0.53, 95% CI 0.36–0.79). A prespecified multivariate analysis, accounting for imbalances in key baseline characteristics, showed a strong OS benefit from the combination (HR = 0.663; 95% CI 0.464–0.947; nominal p = 0.0237) for BRCA1/2 patients (Table 2). More frequent AEs in the experimental arm included anemia (46%), hypertension (31%), constipation (31%), fatigue (26%), nausea (24%), thrombocytopenia (21.2%), and dyspnea (16%). Grade ⩾3 toxicities secondary to abiraterone and niraparib included anemia (28.3%), hypertension (14.6%), and neutropenia (5.2%). Niraparib in combination with abiraterone has been approved by both, FDA and EMA, as first-line therapy for mCRPC patients with BRCA1/2 alterations.
Taken together, the results from these studies suggest a hierarchic benefit aligned with the biology of these tumors, with patients with documented BRCA pathogenic genomic aberrations being the ones more likely to benefit from ARSi and PARPi combinations (Table 2). Although patients with certain non-BRCA HRR pathogenic genomic aberrations could also have some benefit from this approach, further assessment of the underlying molecular mechanisms is needed since this is a very heterogeneous group and some alterations (i.e., PALB2) may sensitize tumors to PARPi significantly more than others (i.e., CHEK2). 33 Finally, some patients without detectable HRR alterations may as well benefit from these combinations. Despite the efficacy may be driven by a potential synergy between PARPi and ARPi, it could also be possible that these tumors harbor HRR deficiency unrelated to genomic events (i.e., epigenomic modifications), or that the impairment of other pathways different from HRR also sensitizes cells to PARPi. 34 (Table 2).
PARPi in combination with immune checkpoint inhibitors
Contrary to other solid tumors, immune checkpoint inhibitors (ICI) have limited efficacy in molecularly unselected PCa.35,36 The rationale for developing PARPi plus ICI combinations relies on the genomic instability associated to HRR and other DDR defects, which may lead to neoantigen production and T-cell activation. 37 The accumulation of cytosolic DNA may also activate the innate immune system, mediating adaptative antitumoral responses. 37 Although this preclinical rationale supports the synergistic activity of PARPi and ICI, previous studies have taught us that molecular selection still should be considered in this approach. Indeed, results from previous phase II trials suggest that, again, patients with DDR deleterious driver aberrations have better outcomes in terms of ORR and/or PSA responses compared to all-comer populations. In the phase III Keylink-010 trial, 35 molecularly unselected mCRPC patients were randomly assigned to treatment with pembrolizumab plus olaparib or an ARSi (abiraterone or enzalutamide). The dual primary endpoints were rPFS by blinded independent central review and OS. Pembrolizumab plus olaparib did not significantly improve rPFS or OS, and this study was stopped for futility. Therefore, molecularly selection probably also matters to better select the mCRPC patients more likely to benefit from PARPi plus ICI combinations, and future trials must take this into account to refine patient selection.
PARPi in combination with radioligand drugs
AR axis can be activated by radiation-induced DNA double-strand lesions, resulting in upregulation DDR genes. 38 This explains the biological rationale to combine PARPi, androgen deprivation therapy (ADT), and radiation-based strategies. As previously discussed, ADT promotes cell death by inducing DDR genes downregulation. In this situation of cell stress, the activity of PARP is increased, thereby justifying the synergy between PARPi, ADT and radioligand drugs.
Lutetium-177 (177Lu)-PSMA-617 is a high tumor-specific radioligand that binds to extracellular surface protein PSMA and releases high doses of β radiation. 39 This molecule has already shown promising activity in patients with mCRPC in different scenarios, both alone and in combination with ARSi.40,41 With regards to PARPi combinations, the phase I LuPARP trial 42 is currently testing the safety of olaparib and (177Lu)-PSMA-617 in mCRPC men.
In a similar way, Radium-223 dichloride is a targeting alpha emitter, that binds areas of high turnover in the bones, such as bone metastases. By releasing high-energy alpha particles, this molecule causes double-stranded DNA breaks and cell death. The NiraRad 43 and COMRADE 44 early-phase clinical trials have tested the safety profile or niraparib and olaparib in combination with radium-223, respectively, concluding that there are no concerning toxicities and these combinations could be further investigated.
PARPi in combination with other agents
Several clinical trials are testing the addition of other targeted therapies to PARPi. As an example, the NCT03840200 phase Ib and NCT02893917 phase II clinical trials have evaluated the combination of rucaparib and the AKT-inhibitor ipatasertib, 45 and the combination of olaparib and the pan-vascular endothelial growth factor receptors tyrosine kinase inhibitor cediranib. 46 The first trial has demonstrated that the combination of ipatasertib and rucaparib is safe but does not have additive antitumor activity, 45 while cediranib combined with olaparib improved rPFS compared with olaparib alone in patients with HRR deficiency but with a significant increase in G3/4 AEs. 46 Prospective exploratory analysis from these and other studies testing the combination of PARPi plus other target-specific drugs could provide more information about underlying mechanisms of resistance, together with a better characterization of biomarkers potentially associated with a better response to these therapies.
Mechanism of resistance to PARPi
Despite the significant rates of initial responses, most mCRPC patients with DDR deleterious genomic aberrations will eventually become resistant to treatment with PARPi. So far, four main mechanisms of PARPi resistance have been described 47 (Figure 2).
(i) Restoration of functional HRR: This is one of the most well-described mechanisms of resistance. Cancer cells can shift from an HRR deficient to proficient status through secondary reversion mutations that restore the open reading frame of those genes, allowing the complete transcription of HRR genes, and, therefore, block PARPi-induced synthetic lethality by regaining their function. Reversion mutations have been identified in multiple PARPi-treated tumors, including prostate cancer.48,49 Demethylation of BRCA1/2 has been reported to restore the gene function in breast cancers. 50
(ii) PARP trapping prevention: PARP trapping to DNA SSB and stalled replication fork formation is key to PARP-induced cell death. Therefore, mutations in PARP which inhibit DNA trapping, or interactions between trapped PARP and other SSB modulators which prevent PARP–PARPi linking lead to drug resistance. 51
(iii) Replication fork stabilization: As previously discussed, stalled replication forks are key mediators in PARPi-induced synthetic lethality. This process is at least in part dependent on the recruitment of nucleases which act in unprotected HRR-deficient stalled replication forks to promote cell death. For that reason, dysregulations in the recruitment of those nucleases to stalled replication forks induce their stabilization, prompting PARPi resistance. 47
(iv) Increase of drug efflux: the drug intracellular uptake can be reduced through upregulation of transmembrane efflux pump proteins such as ABCB1. In these situations, PARPi availability to interact with PARP will be reduced, therefore promoting cell survival and drug resistance. ABCB1 expression has been shown to be correlated with resistance to olaparib and rucaparib in ovarian cancer cell lines. 52

Mechanisms of resistance to PARPi.
There is a gap of knowledge to advise the best management to reverse PARPi resistance. In vitro studies have demonstrated that PARPi and platinum-resistant cancer specimens may be re-sensitized to PARPi when combined with replication stress modulators and cell-cycle checkpoints, among other cell targets. 53 In this setting, ongoing trials are showing promising results when combining PARPi with ATR inhibitors. As an example, the phase II TRAP trial is comparing the responses of PARPi naïve, DDR deficient mCRPC patients with progressive disease after one line of therapy in the metastatic castration-resistant setting and/or one ARSi in the castration-sensitive setting to molecularly unselected mCRPC patients when treated with olaparib plus the ATR inhibitor AZD6738.
Toxicity of PARPi
Despite the above-specified encouraging results PARPi-induced AEs must be considered. Indeed, the most frequent AEs secondary to these drugs include fatigue, and hematological and gastrointestinal toxicities, normally arising within the first 4 months of treatment. 54 Increased PARP trapping has been associated with higher myelosuppression. 55 Preclinical experiments have suggested that PARP1 inhibition is able to induce synthetic lethality in BRCA mutations on its own 10 whilstPARP2 is key for the survival of hematopoietic and stem cells 56 and its inhibition could partially explain the hematological toxicity secondary to these drugs. PARP1-specific inhibitors are being developed with the potential to retain a similar antitumor activity but with a more favorable safety profile. 57
Most of the hematological AEs reported in prostate cancer correspond to acute toxicity, however, PARPi have been associated to an increased risk of myelodysplastic syndrome and acute myeloid leukemia, with a latency period of 18 months since first exposure to PARPi. 58 Only a few cases of myelodysplastic syndromes have been reported in the context of the clinical trials conducted in mCRPC to date, but there is concern that as these therapies are being investigated in earlier stages with more prolonged treatment periods and extended follow-up, the incidence of myelodysplastic disorders might rise. 34
Importantly, no differences in AEs have been noted by HRR status in clinical trials investigating PARPi either alone or in combination4,27–32 therefore careful reconsideration of the potential benefits and side effects is mandatory to limit toxicity when selecting the patients more likely to benefit from these approaches.
Future directions for the development of PARPi in prostate cancer
Despite their attractive targeted mechanism of action, PARPi implementation in daily clinical practice is being limited by the current low germline and somatic testing rates in mCRPC. Increased awareness and access to genomic molecular characterization are mandatory to further personalize the treatment and improve the clinical outcomes of these patients.
When looking at the trial populations from PROPEL, TALAPRO-2, and MAGNITUDE, the great majority of their patients were ARSi naïve. However, real-world mCRPC patients are receiving ARSi earlier and earlier after their diagnosis, either in the mHSPC or non-metastatic castration-resistant (nmCRPC) scenario. Furthermore, multiple studies are currently addressing the benefit of ARSi in earlier stages and the number of patients exposed to ARSi prior to mCRPC is likely to grow in the future. The efficacy of combining ARSi and PARPi is yet to be established in this setting, but the limited responses of treatment with an ARSi after progression to a prior one results reported in previous studies must be considered.59,60 Currently, there is no data supporting that PARPi could re-sensitize tumors to ARSi in these patients. Only the ongoing CASPAR trial (NCT04455750) has allowed prior ARSi for non-metastatic mCRPC, but its results have not been communicated. Moreover, exploratory analysis in the MAGNITUDE trial reported decreased antitumor efficacy in patients who started abiraterone ⩾2–4 months before niraparib. 61
Ongoing trials are investigating the benefit of combining ARSi and PARPi in early stages. In the mHSPC setting AMPLITUDE (NCT04497844) and TALAPRO-3 (NCT04821622) are evaluating the efficacy of the combination of niraparib plus abiraterone and talazoparib plus enzalutamide as first-line therapy in HRR-deficient mHSPC patients. Also in mHSCP, a third study, EvoPAR-PR01 (NTC06120491), is evaluating PARP1 specific inhibitor (seruparib) in combination with abiraterone, enzalutamide or darolutamide in two cohorts of patients with and without HRR alterations, respectively. Other studies are being conducted to assess the effect of PARPi in localized prostate cancer (i.e., NCT04030559, NCT02324998, NCT03570476, NCT05938270). NCT03047135 has investigated olaparib without ADT for patients with high-risk biochemical relapse. All patients with BRCA alterations (n = 11) achieved a PSA decline > 50%. Median PSA PFS was 22 and 13 months in HRR-altered (n = 27) and HRR-proficient (n = 24) patients, respectively. In addition, the TRIUMPH trial (NCT03413995) tested the clinical activity of rucaparib monotherapy as an alternative to frontline ADT or other hormonal therapies in mHSPC patients with germline HRR pathogenic genomic aberrations. Although some prolonged radiological responses were observed, the prespecified threshold of biochemical response for this trial was not met and the trial did not proceed to its second stage.
Despite the great antitumor activity of PARPi in patients with BRCA1/2 and some HRR alterations, resistance eventually arises and the most appropriate subsequent therapy for these patients is unclear. The phase I/IIa trial PETRA has evaluated the antitumor activity of saruparib, a novel PARP1-specific inhibitor, in patients with BRCA1/2, PALB2, or RAD51C/D mutations and different tumor types, including prostate cancer. Patients previously treated with PARPi were eligible (44 out of 61) and responses were noted independently or prior PARPi use. PARPi in combination with ATR or Wee1 inhibition among other targets has signals of molecular activity in some early-phase clinical trials. Other targets like CHK1/2, DNA-PK, ATM, and POLθ, could also become potential combination partners for PARPi in future. 62
Finally, further assessment of long-term toxicity will be paramount to establish the best scenario and duration of treatment with PARPi in prostate cancer.