
Abstract
We present the case of a 31-year-old single male patient, who was admitted through emergency unit with painless hard nodule of his left testis of 6 months’ duration. Ultrasound scan of the scrotum showed a fairly well-defined hypo echoic area in the left testicular parenchyma in its middle part, measuring approximately 10 × 9 mm in size. We performed left inguinal radical orchidectomy. Histopathology examination of the left testis revealed sclerosing Sertoli cell tumor (SSCT) of the testis. This is a very rare testicular tumor with very few published case reports. Systemic examination was performed to exclude systemic metastasis. SSCT is characterized by the presence and aggregates of tubules of Sertoli cells, separated by a sclerotic intercellular matrix formed by fibrotic connective tissue.
Introduction
Sclerosing Sertoli cell tumor (SSCT) of the testis is a sex-cord stromal tumor and is an extremely rare variant of Sertoli cell tumors. Less than 50 cases have been reported in the literature. 1 The first case was described by Zuckerberg et al. in 1991.2,3 This variant has always been described in post-pubertal males and no pre-pubertal cases have been ever reported. The age at onset of SSCT varies between 18 and 80 years, with a mean age of 35 years.
Most patients present with a small, hard, and painless testicular nodule; however, some patients may complain of testicular pain. The testicular nodule is always single and monoliteral, and none of the cases presented describe a hereditary transmission. The tumor is not associated with extragonadal clinical signs and symptoms like gynecomastia and accelerated bone growth due to hormone production or dysplastic syndromes.2–4
Ultrasound (US) findings do not differentiate between SSCT and other variants like Sertoli cell tumors not otherwise specified (NOS) or other germinal testicular tumors.
SSCT appear as small, well demarcated and hard lesions in the testicular parenchyma. 2 Since Sertoli tumors are benign, the standard treatment can be considered a testis-sparing surgery in small lesions (less than 15 mm). However, this approach should be strongly recommended in patients with small bilateral lesions, monorchid or men with multiple neoplasia syndrome or familiar cases, and even in lesions with hyperechoic appearance on US suggesting large-cell calcification. A definitive diagnosis can be made only by histopathological examination.
Case report
A 31-year-old single male patient presented at the accident and emergency of Al Khor Hospital in Qatar complaining of a painless hard nodule of his left hemi-scrotum of 6 months’ duration. He reported a history of trauma of the left scrotum in his childhood while playing cricket. The patient has no history of hypertension, diabetes mellitus, lower urinary tract symptoms, or urethral discharge. He denied any sexual contact.
Complete blood tests and renal function tests were normal, as were the following tumor markers: ALPHA- FETO protein 3, BCHG < 1, LDH 177. Hormone tests [luteinizing hormone (LH), follicle-stimulating hormone (FSH), total testosterone] were also normal.
US and computed tomography findings
US of scrotum showed both testicles to be normal in size and echotexture. There was a fairly well-defined hypo echoic area in the left testicular parenchyma in its middle part, measuring approximately 10 × 9 mm in size (Figure 1).

US of left testis.
Computed tomography (CT) of the abdomen, pelvis, and thorax was conducted with contrast. No obvious mass lesions or lymphadenopathy were detected. The patient underwent left inguinal radical orchidectomy.
Histopathology
Macroscopic findings
The cut surface of the testis shows a circumscribed grey white firm area measuring 1.0 × 0.8 × 0.8 cm towards the hilar region. The surrounding normal testis appears unremarkable.
Microscopic description
Sections show a sex-cord-stromal tumor that is attached to the tunica and characterized by tubules, cords, and solid clusters of medium-sized polygonal cells with abundant eosinophilic cytoplasm, ovoid nuclei, and conspicuous, and sometimes prominent, nucleoli. Immunohistochemical (IHC) analysi showed that the tumor cells are positive only with synaptophysin and WT–1, while negative with OCT3/4, (D2–40), Melan-A, HMBE-1, CK AE1/AE3, SALL-4, Inhibin, Calretinin, CK 7, CK 20, Ber-EP4, CD34, CD31, CK 5/6, PSA, Cdx-2, and CEA. Ki-67 is less than 1%. (Figures 2–6)
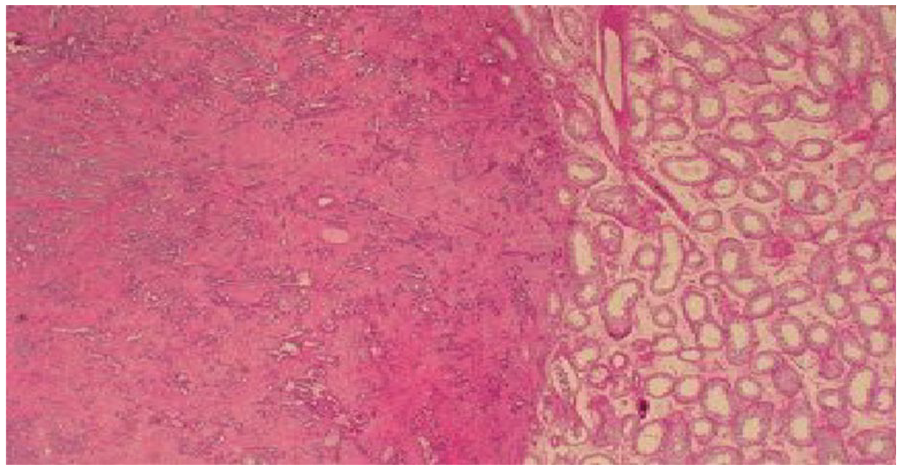
Normal testicular tissue.

Normal testicular tissue.
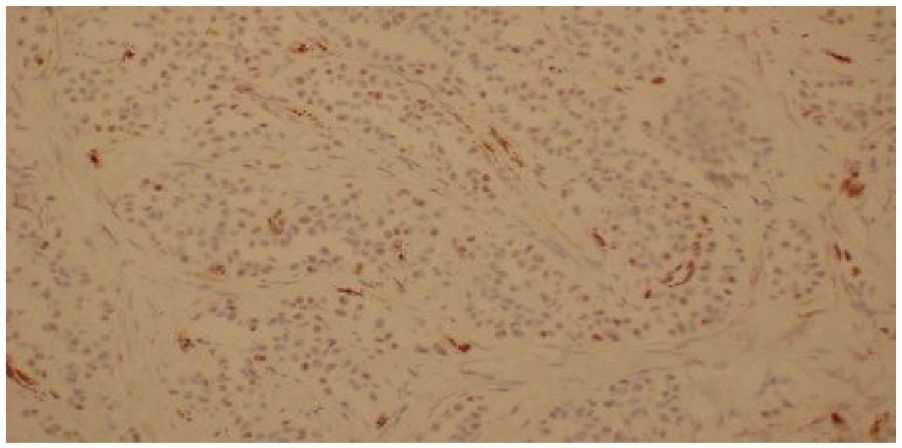
Tumor cord.

Synaptophysin positive.

WT1 positive.
The post-operative course was uneventful, and, after 6 months of follow up, no recurrence, no lymph node involvement or distant metastasis has been observed.
Discussion
SSCT of the testis is a sex-cord stromal tumor composed of varying degree of features of fetal, prepubertal, or adult Sertoli cells. 5 This is a rare tumor, accounting for less than 1% of all testicular tumors, and is classified by the World Health Organization (WHO Classification 2004) into three histological subtypes: Sertoli cell tumor NOS, large cell calcifying Sertoli cell tumor, and SSCT. 5
SSCT is very rare variant of Sertoli cell tumors; worldwide, less than 50 cases have been reported in the literature. 8 First described by Zuckerberg et al. in 1991,2,7 this variant has always been described in post-pubertal males and no cases have been ever reported in pre-pubertal males.
Age at onset of SSCT varies between 18 and 80 years, with a mean age of 35 years.
Since Sertoli tumors are benign, the standard treatment can be considered a testis-sparing surgery in small lesions (less than 15mm). However, this approach should be strongly recommended in patients with small bilateral lesions, monorchid or men with multiple neoplasia syndrome or familiar cases, and even in lesions with hyperechoic appearance on US, suggesting large-cell calcification.
Most patients present with small, hard, and painless testicular nodule; however, some patients may complain of testicular pain. The testicular nodule is always single and unilateral, and no hereditary transmission was presented or described in these cases.
Patients do not present with extra gonadal clinical signs or symptoms such as gynecomastia and accelerated bone growth due to hormone production or dysplastic.2,3,4
Sex cord stromal tumor should be considered in the case of small testicular masses where there are hormone disorders or normal tumor markers. Immediate orchiectomy should be avoided, favoring testis-sparing surgery. 7
Small Leydig cell tumors usually present as well-defined homogeneously hypoechoic lesions, often located peripherally in the testis, with prominent vascularization upon color Doppler interrogation. However, large-cell calcifying Sertoli cell tumors are usually echogenic masses with large areas of calcifications, or extensively calcified lesions. Non-calcifying Sertoli cell tumors (classic and sclerosing histotype) are typically hypoechoic, well-circumscribed, with round-to-lobulated masses. The ultra-sonographic appearance of the other histotypes is not specific. 8
Most of these tumors are small (less than 2 cm in diameter); however, some lesions are large and lesions exceeding 7 cm have been reported. 9 Lesions appear as small, well demarcated, and hard in the testicular parenchyma. 2
The color of the lesion appears from white to a yellowish–brown. Histopathological examination is the only way for a definitive diagnosis to be made.
The histopathology of SSCT is characterized by the presence and aggregates of tubules of Sertoli cells, separated by sclerotic intercellular matrix formed by fibrotic connective tissue. The cytoplasm is small, with pale coloration and contains large lipid vacuoles; however, no calcifications are present. Nuclei are round in shape and have different sizes. 2
IHC examination of SSCT is characterized by positive synaptophysin, vimentin, and CD56, while chromogranin A is negative.2,10 The expression of cytokeratin and inhibin alpha is negative.
SSCT is treated by high inguinal radical orchiectomy; however, the optimal treatment for this variant is enucleation of the lesion and preservation of the testicular tissue. Since the intraoperative frozen section is not always available, or reliable, depending on pathologist experience, Sbrollini et al. suggested that the nodule should be excised and additional biopsy of normal-looking tissue performed. Then any radical surgery (orchiectomy) should be considered after definitive histopathological examination, that is the only way for a definitive diagnosis to be made. 11 Galosi et al. were able to save testis in 40% of patients presenting with lesions smaller than 15 mm, discovering benign lesions. 12
Intraoperative diagnosis can be difficult, but with the collaboration of pathologist, the surgeon can safely perform testis-sparing surgery (if this is feasible). In instances of uncertainty at intraoperative analysis, the surgeon makes a decision to conserve the testis and awaits a definitive pathology consultation, based on which the surgeon will undergo a second-look surgery (delayed orchiectomy) or follow up. 13
The prognosis of this tumor is thought to be good because, in most reported cases, there was no tumor recurrence or lymph node or distant metastasis; however, only one case was reported to had lymph vascular invasion and invasive growth and distant metastasis to bone.
Conclusion
SSCT is a very rare testicular tumor seen in post pubertal males. The majority of patients present with single and monoliteral small, hard, and painless testicular nodule. Intraoperative diagnosis can be difficult, but with the collaboration of a pathologist, the surgeon can safely perform testis-sparing surgery (if this is feasible) or a second look surgery (delayed orchiectomy). SSCT has a good prognosis as the majority of the cases showed no tumor recurrence or distant metastasis.
Footnotes
Author Contributions
FSG researched literature and conceived the study. FSG was involved in protocol development, gaining ethical approval, patient recruitment and data analysis. FSG wrote the first draft of the manuscript. FSG reviewed and edited the manuscript and approved the final version of the manuscript.
KHB, NS, AMS, AAY, KMA, AHS, WSB and NAC reviewed the manuscript, helped in finalizing it and approved the final version of the manuscript.
HA studied and reported the histopathology specimen.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Ethical approval
The ethics committee of Hamad Medical Corporation approved this study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed consent
A written informed consent was obtained from the patient to publish his medical data and images. Consent approved from ethical and research committees of institute obtained.
Guarantor
FSG