
Abstract
Hereditary spastic paraplegia (HSP) groups rare, clinically and genetically heterogeneous neurodegenerative disorders, characterized by progressive lower-limb spasticity and weakness. Over the past decades, diagnostic strategies have evolved from pure clinical assessment to the integration of molecular tools, with next-generation and long-read sequencing (LRS) substantially increasing diagnostic yield and refining genotype–phenotype correlations. Neuroimaging provides complementary information, especially in specific subtypes such as SPG11 and SPG15, supporting diagnosis and guiding testing. Treatment has historically focused on symptomatic care, including physiotherapy, antispastic agents, and botulinum toxin, with dalfampridine explored for gait improvement in selected patients. More recently, research has expanded into disease-modifying avenues, such as drug repurposing (e.g., statins in SPG5, menatetrenone in ALS2) and early gene-based interventions in ultra-rare subtypes. At the same time, advances in technology, ranging from quantitative imaging and digital gait analysis to induced pluripotent stem cell models and artificial intelligence, are beginning to influence both clinical management and trial design. This review traces the trajectory of HSP care from its historical foundations to present standards and emerging innovations, outlining how technological progress is shaping realistic prospects for future therapeutic strategies.
Plain language summary
Hereditary spastic paraplegia (HSP) refers to a group of rare neurological conditions that mainly affect the legs, causing stiffness (spasticity), weakness, and difficulty walking. For many years, treatment options were limited, focusing mainly on easing symptoms through physical therapy or medications that reduce muscle stiffness. Genetic testing was not widely available, and many people went undiagnosed. In recent years, science and medicine have made major progress in understanding and managing HSP. Thanks to advanced genetic technologies, doctors can now identify the specific gene changes that cause the condition in many patients. This has opened the door to more targeted and personalized treatments. For example, gene therapy and antisense oligonucleotides – two forms of genetic medicine – are being tested in clinical trials and have already shown promising results in some rare forms of HSP. In parallel, researchers are finding new ways to repurpose existing drugs to treat HSP more quickly and effectively. Digital health tools like wearable sensors and smartphone apps are also helping patients and doctors to track symptoms and responses to treatment in real time, even from home. This article explains how treatment for HSP is moving from a “one-size-fits-all” approach to more personalized care, tailored to each person’s unique genetic and clinical profile. It also highlights how new technologies—like artificial intelligence—are helping scientists understand disease better and design more precise therapies. Although many treatments are still under development, the latest breakthroughs are offering new hope for individuals and families living with HSP.
Keywords
Introduction
The term hereditary spastic paraplegia (HSP) refers to a clinically and genetically heterogeneous group of inherited neurodegenerative disorders, primarily affecting the long motor neurons of the corticospinal tract. 1 The hallmark clinical features, due to axonal degeneration of the longest descending motor pathways, are progressive lower limb spasticity, muscle weakness, and hyperreflexia, often accompanied by bladder dysfunction and gait abnormalities. 2 HSP is traditionally categorized into two main forms: pure forms, where motor impairment is restricted to the lower limbs, and complex forms, where spastic paraparesis is accompanied by additional neurological or systemic features. These may include ataxia, peripheral neuropathy, muscle atrophy, seizures, dementia, intellectual disability, and extrapyramidal signs, reflecting involvement of neural systems beyond the corticospinal tracts. While this dichotomy remains clinically useful, many patients show intermediate or overlapping phenotypes, underscoring the wide spectrum of presentations observed in clinical practice. 3
Epidemiologically, HSP affects an estimated 0.1 to 5 individuals per 100,000 globally. Its prevalence varies depending on the mode of inheritance, presence of founder effects, and level of consanguinity.4,5 The age at onset, ranging from infancy to late adulthood, is highly variable even within the same genetic subtype, further contributing to the broad clinical spectrum. 6
The genetic landscape of HSP is notably complex, with over 80 genes implicated in the disease.1,2,7,8 These genes encode proteins involved in various cellular processes, including axonal transport, mitochondrial function, lipid metabolism, and membrane trafficking. 2
Importantly, mutations in different genes may converge on similar clinical phenotypes, and conversely, identical mutations may lead to variable presentations, even within the same family. 2 Autosomal dominant forms account for the majority of cases in Western countries, with SPAST (spastic paraplegia gene 4 (SPG4)), ATL1 (SPG3A), REEP1 (SPG31), and KIF5A (SPG10) among the most frequent causes. Autosomal recessive forms, however, show remarkable genetic heterogeneity and are enriched in populations with high consanguinity, with SPG11 and SPG15 being among the most common worldwide. In addition, rarer X-linked and mitochondrial patterns of inheritance further expand the complexity of these conditions. 9 The clinical and genetic variability poses significant challenges for diagnosis, classification, and management, and underscores the need for individualized approaches in both clinical care and research. 2
Recent years have brought rapid advances in areas such as healthcare technology, medical informatics, and computational sciences, significantly enhancing our ability to diagnose, monitor, and tailor treatments for patients with HSP. These developments have not only improved current standards of care but also opened up new possibilities for precision medicine and patient-specific therapeutic strategies. The integration of advanced imaging, next-generation sequencing (NGS), machine learning, and bioengineering is gradually supporting earlier and more accurate diagnosis and informing patient management, enabling a more accurate understanding of disease mechanisms and identifying candidate targets for intervention.
In the present work, HSPs are considered in their entirety, with a focus on general mechanisms, diagnostic strategies, and therapeutic approaches that are relevant across the spectrum. Specific subtypes (such as SPG4, SPG11, or SPG7) are mentioned only when they provide illustrative examples, clarify genotype–phenotype correlations, or highlight data from studies conducted in defined cohorts.
This review traces the evolution of diagnostics and treatment in HSP over time, moving from the recent past to today’s innovation-driven landscape and the future therapeutic promise offered by the ongoing progress across multiple fields—genetics, laboratory diagnostics, computational sciences, bioinformatics, and genetic engineering.
The past: from clinical observation to symptom management
Diagnostic limitations of the pre-genomic era
In the pre-genomic era, diagnosis of HSP was based primarily on clinical observation and family history, with minimal access to genetic testing and virtually no molecular characterization. This phenotype-driven approach, while useful for recognizing core clinical patterns, resulted in a fragmented understanding of the disease spectrum and often failed to clearly distinguish between subtypes. The lack of molecular insight limited diagnostic accuracy and also hindered progress in unraveling the underlying pathophysiological mechanisms of the disease. Therapeutic strategies were therefore confined to symptom management, with interventions aimed at alleviating spasticity, mobility issues, or secondary complications. The absence of genotype-based stratification precluded the development of precision medicine approaches and disease-modifying therapies, reinforcing a model of care that addressed symptoms rather than causes.
The following paragraph summarizes the symptomatic approaches previously and still adopted in HSP—both in genetically characterized cases and in many others diagnosed solely on clinical grounds.
Traditional symptomatic approaches (medications, physical therapy, surgical procedures)
Pharmacological treatment for HSP remains largely symptomatic, although several drugs have been evaluated for their potential benefits in selected subtypes or in the presence of certain clinical features. Gabapentin, an antiepileptic agent that acts via inhibition of calcium channels, was tested in a small crossover trial in SPG4 patients but failed to demonstrate statistically significant differences versus placebo. 10 In another trial, the GABA (gamma-aminobutyric acid) receptor agonist progabide reduced spastic hypertonia and reflex activity; however, its relevance for HSP remains unclear as the treatment was short-term, and the outcomes lacked clinical significance. 11 Dalfampridine (4-aminopyridine), a potassium channel blocker used in multiple sclerosis, has been tested in two open-label studies and in an observational series of patients with different HSP genotypes or clinical diagnoses.12,13 These studies reported improvements in gait parameters, motor function, and fatigue, although all were limited by small sample sizes and heterogeneity of included patients.
L-Dopa has been tested in cases of HSP with overlapping Parkinsonian features,14–18 specifically SPG11 and one case of SPG8. While some patients showed partial improvement in motor symptoms, the available data are anecdotal, based on uncontrolled observations, often lacking standardized outcome measures. Nonetheless, in rare cases of complex HSP with dopaminergic involvement, L-Dopa may be considered. A more systematic investigation was carried out in a prospective cohort of 22 patients with SPG11, where DAT-SPECT imaging confirmed widespread nigrostriatal degeneration but a single-blind trial using L-Dopa failed to show significant clinical benefit. 19 Despite the many limitations recognized by the authors, this study suggested that dopaminergic degeneration is a consistent feature in SPG11 but that postsynaptic mechanisms may limit clinical response to L-Dopa treatment. We cannot exclude clinical benefits occurring in selected patients presenting parkinsonian features.
Intramuscular botulinum toxin type A (BoNTA) is widely used for focal spasticity management. In HSP, both uncontrolled, randomized, open-label, and retrospective studies have been conducted.20–23 One uncontrolled trial showed a significant increase in comfortable gait velocity and a transient reduction in muscle tone after injections in each triceps surae, although strength and balance remained unchanged. 18 Other studies report that the use of BoNTA, often in association with physical therapy, showed modest improvements in spasticity measures over time.24,25 The largest randomized, placebo-controlled crossover trial to date (SPASTOX) confirmed the safety of BoNT-A and demonstrated a reduction in adductor tone, but failed to show significant improvements in gait or other functional outcomes. 21 While limited by the injection protocol, dosing, and outcome measures, this study represents the highest level of evidence currently available and underscores that spasticity reduction alone may not translate into meaningful functional benefit in HSP.
A more mechanism-based approach has been explored in SPG5, where biallelic mutations in CYP7B1 lead to neurotoxic oxysterol accumulation. Cholesterol-lowering drugs such as atorvastatin, simvastatin, and ezetimibe have shown consistent reductions in 27-hydroxycholesterol, a validated SPG5 biomarker.26–28 However, no study to date has shown significant improvements in motor or functional parameters, and the longer-term effects and clinical benefit remain uncertain. A more recent phase II crossover trial supported the biochemical efficacy of atorvastatin, chenodeoxycholic acid, and resveratrol, albeit again without clinical outcome evaluation.
Finally, in ultra-rare cases of HSP linked to methylenetetrahydrofolate reductase deficiency, supplementation with betaine, folinic acid, and vitamins led to biochemical correction of hyperhomocysteinemia and, in some patients, a subjective or modest clinical improvement.29,30 Given the very low quality of the available evidence, these approaches are currently limited to highly specific metabolic subtypes. Table 1 lists the medications used to date in HSP together with the dosages adopted in clinical studies.
Drug regimens studied to date in HSP.

Patient cohorts were heterogeneous, including both clinically defined HSP and genetically confirmed cases, with various SPG subtypes represented (e.g., SPG4, SPG7, SPG11).
HSP, hereditary spastic paraplegia; L-Dopa, levodopa; MU, mouse units; SPG, spastic paraplegia gene.
The therapeutic role of physical and surgical interventions in HSP remains inadequately characterized, with most available data stemming from case reports and low-quality uncontrolled studies. Although physical therapy is commonly recommended, robust evidence supporting its use is lacking, as are clear guidelines regarding the optimal regimen or timing. Similarly, electrical stimulation has been explored only in a single patient, aged 26, who showed a 27% increase in gait velocity following 3 months of bilateral stimulation of the quadriceps and anterior compartment muscles. 31
Robot-assisted gait training has also been evaluated in HSP. One uncontrolled trial using the Lokomat system in 13 patients with pure HSP found statistically significant improvements in balance and walking speed after 6 weeks of training, with benefits maintained at 2-month follow-up. 32 Hydrotherapy was examined in a small uncontrolled trial of nine patients, who reported significant changes in gait kinematics and kinetics after a 10-week intervention. 33 Interestingly, rather than correcting pathological gait patterns, hydrotherapy appeared to reinforce existing compensatory mechanisms.
Among the surgical options, intrathecal baclofen has shown promise in reducing spasticity and improving gait in patients unresponsive to oral antispastic agents. One open-label study of 14 adults demonstrated statistically significant improvements in Modified Ashworth Scale scores and walking ability over a 25.8-month follow-up. 34 Selective dorsal rhizotomy (SDR), traditionally used in cerebral palsy, has been trialed in both adults and children with HSP. Four adults obtained long-term improvements in spasticity and spasm frequency without complications, 35 while a retrospective study of four pediatric cases, including patients with mutations in ALS2, showed stable spasticity reduction but progression of functional decline in two individuals with complex HSP phenotypes. 36 Overall, while SDR may be a feasible intervention for selected cases of pure HSP, its role in broader clinical practice remains uncertain due to limited and low-quality evidence. Collectively, these findings highlight the urgent need for high-quality controlled studies to define the role of physical and surgical treatments in HSP.
Barriers to clinical translation and future directions
Although numerous pharmacological, physical, and surgical interventions have been tested in HSP, their impact on clinical practice remains limited. Most studies have enrolled small and heterogeneous cohorts, and often mixed different genotypes or combined pure and complex forms, which makes it difficult to generalize the results. Methodological weaknesses, including uncontrolled or open-label designs, short observation periods, and the use of outcome measures that are not sufficiently sensitive or anchored to patients’ needs, have further restricted the ability to demonstrate meaningful benefits. The lack of validated biomarkers and standardized trial protocols has also hampered reproducibility, while the rarity of HSP and the fragmentation of patient populations across centers have posed significant logistical challenges for recruitment and trial implementation. This has been clearly illustrated by a recent scoping review, which systematically examined outcome measures and biomarkers applied in HSP research and reported marked heterogeneity across studies, underscoring the urgent need for a standardized core outcome set to enable more robust and comparable clinical trials. 37
To overcome these barriers, several strategies should be considered. Grouping patients according to genotype or clinical profile, or pooling HSP cohorts with other spastic disorders, could create larger and more homogeneous populations suitable for interventional studies. The establishment of multicenter and international networks will be essential to ensure adequate recruitment and methodological harmonization. Recent initiatives illustrate that this is feasible: the PROSPAX consortium (https://www.prospax.net/) has launched longitudinal studies in SPG7 and ARSACS, combining digital gait analysis, MRI, fluid biomarkers, and patient-centered outcomes to generate trial-ready measures; the ERN-RND (https://www.ern-rnd.eu/) provides a European infrastructure for shared registries and harmonized protocols across rare neurological disorders; and specific tools such as the TreatHSP-QoL have been developed to capture patient-reported outcomes relevant to everyday life in HSP. 38 In parallel, broader registries such as the ARCA Registry and STOP-HSP.net have demonstrated the feasibility of multinational, collaborative natural history studies.39,40
Equally important is the adoption of more sensitive and standardized outcome measures, including digital gait analysis, wearable sensors, imaging markers, and fluid biomarkers, which may capture treatment effects missed by conventional clinical scales. In this regard, the role of regulatory agencies is crucial. Both the European Medicines Agency (https://www.ema.europa.eu/en/qualification-novel-methodologies-medicine-development) and the US Food and Drug Administration (https://force-dsc.my.site.com/ddt/s/) are increasingly promoting the development, validation, and harmonization of biomarkers and digital endpoints through dedicated qualification programs. The alignment of these policies with multicenter research efforts will be essential to ensure that outcome measures developed in rare diseases like HSP can be recognized internationally, thereby facilitating trial design, drug approval, and ultimately the translation of research findings into clinical practice.
The present: toward precision medicine and targeted approaches
Improving understanding of the pathophysiology of hereditary spastic paraplegia
In recent years, understanding of the pathological mechanisms underlying HSP has significantly improved thanks to advances in diagnostic technologies, the development of sophisticated preclinical models, and the integration of multi-omics data (Table 2). Together, these approaches have provided deeper insights into the biology and genetic complexity of the disease. The following sections explore these technological advances, showing how they have enhanced knowledge of HSP pathogenesis and facilitated the development of targeted therapeutic strategies. Figure 1 summarizes the advances in diagnostics enabled by LRS and technological integration, the improved computational capacity fostering integration between multi-omics and in vivo/in vitro modeling, and current therapeutic developments for hereditary spastic paraplegia, specifically gene therapy and drug repurposing.
Recent diagnostic advances and their implications for HSP.

HSP, hereditary spastic paraplegia.

From diagnosis to therapy: the current state of precision medicine.
Advances in diagnosis
The advent of NGS technologies has revolutionized the HSP diagnostic landscape.7,41,42 Unlike traditional Sanger sequencing methods, used to analyze a limited number of candidate genes, NGS enables the simultaneous evaluation of hundreds of genes, significantly reducing diagnostic times and costs. 43 The implementation of whole-exome sequencing (ES) and, more recently, whole-genome sequencing has further expanded the scope of genetic analysis, making it possible to identify both coding and non-coding pathogenic variants. Additionally, LRS, a cutting-edge technology capable of producing DNA reads longer than 10 kb, has enabled more accurate detection of structural variants, repetitive regions, and complex genomic rearrangements that are often missed by short-read methods.44,45 In the context of HSP, LRS is a powerful tool for uncovering pathogenic variants in patients who remain undiagnosed after ES. The extended read lengths of LRS allow comprehensive analysis of entire genes and can capture deletions, insertions, or duplications across multiple exons in a single read. In a recent study, researchers applied LRS to seven ES-negative HSP cases and successfully identified intragenic deletions in SPAST or PSEN1 in all five families analyzed. 46
This major technological development, enabling the discovery of novel disease-associated genes and previously undetected rare variants, has led to a marked increase in genetic diagnoses of HSP. Moreover, the growing catalog of molecular findings has paved the way for a molecular reclassification of HSP, moving beyond the traditional phenotype-driven taxonomy toward a genotype-based framework. 47 This refined stratification, grounded in genotype–phenotype correlations, is not only enhancing understanding of the disease mechanisms 2 but also laying the foundations for precision medicine approaches, including patient-specific prognostics and the design of targeted therapeutic interventions.
In parallel with these advances in molecular diagnostics, neuroimaging remains an essential complementary tool in the evaluation of HSP. Conventional MRI can reveal characteristic features that support the clinical diagnosis and guide genetic testing, such as a thin corpus callosum and the “ears of the lynx” sign in SPG11 and SPG15, or cerebellar atrophy in SPG7. 48 Beyond these qualitative cues, advanced MRI techniques, including diffusion tensor imaging and volumetric analyses, have demonstrated widespread microstructural abnormalities and subcortical changes across different subtypes, highlighting their potential as biomarkers of disease progression.49,50 Magnetic resonance spectroscopy has further identified metabolite alterations, such as reduced N-acetylaspartate and increased lipids in specific forms like SPG11 and SPG54, which may correlate with clinical severity and neuronal dysfunction. 51 More recent multimodal approaches underline how quantitative imaging can capture subtle changes invisible on routine MRI, strengthening its role not only in diagnostic workflows but also as a translational research tool with direct implications for clinical trial design.49,50
Development of preclinical models
The development of preclinical models of HSP has encountered significant challenges that limit their relevance to human pathology. Murine models, for example, frequently failed to reproduce the full clinical spectrum, often exhibiting normal survival rates and only subtle or late-onset motor deficits. 52 Similarly, zebrafish models, although invaluable for high-throughput genetic studies and early neurodevelopmental analyses, inherently lack a corticospinal tract, the principal neural pathway affected in HSP. 53 Recent technological advances have facilitated a shift from traditional two-dimensional culture systems to increasingly complex three-dimensional structures derived from induced pluripotent stem cells (iPSCs). Ranging from brain organoids to cortico-spinal assembloids capable of reconstructing entire motor circuits, these iPSC-derived models more faithfully recapitulate the cytoarchitecture and cellular heterogeneity of human neural tissues, offering unprecedented opportunities to model disease processes at the single-patient level. The application of iPSC technologies, elucidating key mechanisms such as axonal degeneration, mitochondrial dysfunction, and defects in endolysosomal trafficking, has notably advanced understanding of the molecular and cellular pathophysiology of several HSP subtypes, including SPG3A, SPG4, SPG5, SPG7, SPG11, SPG15, SPG47, SPG48, SPG50, SPG51, SPG52, and SPG57. 54
Despite their potentialities in terms of translational research, iPSC-derived HSP models present several intrinsic limitations that must be carefully considered. In spite of the technological breakthroughs that have led to their generation, these complex, three-dimensional models remain reductionist when compared with the anatomical and functional complexity of the human brain and body. 54 Crucial cell populations, such as microglia, vascular cells, and pericytes, are often absent or poorly represented since they originate and develop outside the neural tube. Efforts to incorporate these elements are ongoing but remain technically challenging. Furthermore, the reprogramming process itself induces profound metabolic and mitochondrial alterations, including a shift toward glycolytic metabolism and significant remodeling of mitochondrial architecture, which may impact disease modeling, particularly of HSP subtypes associated with mitochondrial dysfunction. 55 Furthermore, variability between iPSC clones derived from the same donor complicates experimental reproducibility; strategies such as the use of multiple clones or the generation of isogenic lines through gene editing are increasingly employed to mitigate this issue. In short, iPSC technology offers a powerful tool for advancing understanding and treatment of HSP, but further methodological refinement is needed, and therefore, the data it generates should be interpreted with caution.
Multi-omics
Approaches combining multiple “omics” (transcriptomics, epigenomics, metabolomics, etc.) have become increasingly valuable in the study of rare diseases, which often involve complex and poorly understood molecular mechanisms that cannot be fully captured through single-layer analyses. In the context of HSP, multi-omic approaches appear particularly promising. Transcriptomic analyses help in linking gene expression patterns with clinical phenotypes, while metabolomic and proteomic data can highlight disruptions in energy metabolism and lipid homeostasis. Epigenomic studies may further elucidate how gene regulation contributes to neuronal vulnerability. Together, these strategies could offer new insights into the pathogenesis of HSP and pave the way for precision medicine approaches in its diagnosis and treatment.56–58
Recent transcriptomic studies have begun to unravel disease mechanisms in HSP. Recently, 59 researchers performed large-scale RNA sequencing on Spg11–/– mice across different ages and brain regions, revealing dynamic dysregulation of pathways related to neuroinflammation, RNA metabolism, synaptic development, and cell proliferation. These findings confirm known defects in autophagy–lysosomal pathways and highlight additional mechanisms that may contribute to disease onset and progression.
Metabolomic and lipidomic studies are also providing insights into HSP. In a recent study, 60 patients with SPG35 due to FA2H deletions were investigated using lipidomics on peripheral blood mononuclear cells and demonstrated profound alterations in sphingolipid metabolism, supporting their role as potential biomarkers. Similarly, our group 61 analyzed patients with HPDL-related spastic paraplegia and identified distinctive metabolic profiles affecting mitochondrial function, linking rare genetic variants to measurable bioenergetic defects. A Drosophila model of ATL1/SPG3 62 showed that genetic background and epigenetic regulation can modulate motor phenotypes, providing a proof-of-principle that omic profiling can reveal modifiers of disease severity.
Proteomic strategies have also been applied to HSP, though the field is still emerging. A high-content screening study in AP-4-deficiency incorporated integrated proteomic and transcriptomic analyses to identify compounds that correct ATG9A trafficking defects, demonstrating how proteomics can inform therapeutic development. 63 Martinello et al. 64 summarized proteome-wide patterns across different HSP subtypes, highlighting common pathways involving protein trafficking, mitochondrial function, and lipid metabolism. In another study, 65 protein interaction networks combined with machine learning were used to predict functions of HSP-related proteins, illustrating the potential of proteomic and network-based inference to expand disease knowledge.
By contrast, studies applying epigenomic methods to HSP remain extremely limited. In their work, Candia and co-workers demonstrated in a Drosophila model of ATL1/SPG3 that Polycomb group proteins act as genetic modifiers of disease severity, suggesting that chromatin regulation contributes to phenotypic variability. However, systematic investigations using epigenomic profiling techniques such as DNA methylation analysis, ATAC-seq, or ChIP-seq are still lacking in HSP. Addressing this gap could be crucial, as epigenetic regulation may help explain differences in age at onset, disease progression, and therapeutic responsiveness among patients carrying the same genetic variants. Together, these strategies could offer new insights into the pathogenesis of HSP and pave the way for precision medicine approaches in its diagnosis and treatment.66,67
The gap between precise genotyping and current treatments
Although diagnostic techniques have improved—particularly as a result of multi-omic technologies and functional studies—considerably deepening our understanding of the underlying molecular mechanisms of HSP, a substantial gap persists between this growing pathophysiological knowledge and the availability of effective therapies. The translation of molecular discoveries into clinically actionable treatments remains limited, and for the vast majority of HSP cases, current therapeutic management strategies are still largely restricted to symptomatic relief rather than disease modification.
The following paragraphs provide an overview of ongoing clinical trials in HSP and examine gene therapy, looking at its advances, applications, and limitations. Consideration of potential solutions leads to the subsequent section, which discusses how these therapies are becoming increasingly personalized. We also consider the growing shift toward a greater focus on quality of life rather than mere treatment of the underlying cause, and the integration of technological advances in computational power and artificial intelligence (AI) into these processes.
A snapshot of current clinical research
As shown by a ClinicalTrials.gov search performed in June 2025 using “hereditary spastic paraplegia” as the input term, clinical research in HSP has expanded significantly in recent years, with trials conducted across multiple continents. The United States, China (especially Shanghai), Italy (two centers in particular—IRCCS Eugenio Medea and IRCCS Fondazione Stella Maris), and France emerge as leading hubs. Most trials began between 2023 and 2025, and they indicate a growing interest in both observational and interventional studies. The latter slightly predominate with interventions including pharmacological therapies, robot-assisted physical rehabilitation, and neurocognitive interventions. While some trials focus exclusively on HSP, many also explore spasticity in the broader framework of neurological conditions such as amyotrophic lateral sclerosis (ALS), primary lateral sclerosis (PLS), and spinal cord diseases. SPG4, SPG5, and SPG11 are the most commonly investigated subtypes in trials dedicated specifically to HSP. For example, ongoing and completed trials at Boston Children’s Hospital and IRCCS Fondazione Stella Maris focus on genetic characterization in SPG4 and therapeutic approaches (trehalose or miglustat) in SPG11, respectively. The cohorts in these subtype-specific trials vary in size, typically ranging from 15 to 100 subjects, depending on the rarity of the subtype and the scope of the trial. Supplemental Table 1 lists the main characteristics of the clinical research trials identified. All clinical studies listed in Supplemental Table 1 are identified by their ClinicalTrials.gov registration numbers (NCT identifiers). These identifiers correspond to publicly accessible registry records. The trials included in the analysis are: NCT 06948019, 06936163, 06844734, 06742697, 06728787, 06692712, 06573866, 06572046, 06553976, 06478238, 06260982, 06229626, 05848271, 05767268, 05613114, 05373082, 05518188, 05354622, 05196178, 05174403, 04912609, 04875416, 04768166, 04712812, 04256681, 04180098, 04101643, 03981276, 03961906, 03627416, 03206190, 03204773, 03104088, 02859428, 02852278, 02604186, 02327845, 02314208, 01568658, and 00023075 (all registered on ClinicalTrials.gov; the “NCT” prefix applies to all identifiers).
Gene addition, gene editing, and gene silencing
Gene therapy, aiming to treat or prevent genetic diseases by modifying the genetic material within cells, has emerged as one of the most transformative innovations in modern medicine. 68 Early attempts date back to the 1990s, but a major breakthrough occurred in 2017 with the Food and Drug Administration (FDA) approval of Luxturna for Leber congenital amaurosis, the first directly administered gene therapy in humans. 69 Since then, gene therapy strategies have evolved rapidly, offering new therapeutic possibilities for rare neurogenetic disorders like HSP.
The three main approaches currently available are gene addition, gene editing, and gene silencing.70–73 Gene addition, also known as gene augmentation, involves introducing a functional copy of a gene into target cells to restore or compensate for lost or defective gene function. This is often achieved using viral vectors, such as adeno-associated viruses (AAVs), which deliver the therapeutic gene without integrating it into the host genome. In contrast, gene editing uses engineered nucleases like CRISPR–Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats), transcription activator-like effector nucleases, or zinc finger nucleases to induce targeted changes in the DNA sequence, enabling correction, insertion, or deletion of genetic material at specific genomic loci. These changes are permanent and are typically mediated by cellular DNA repair mechanisms. Finally, gene silencing reduces or abolishes the expression of harmful genes at either messenger RNA (mRNA) or transcriptional level via CRISPR interference. RNA-based therapies aim to directly influence gene expression through the use of RNA molecules, such as antisense oligonucleotides (ASOs), small interfering RNA, and mRNA. The main advantages of these therapies include their ability to target specific genetic mutations, potentially bypassing the limitations of traditional treatments like systemic drugs. Drawbacks, however, include issues with delivery (especially to the central nervous system) and RNA stability, and the risk of unintended side effects due to the modulation of genes crucial for other cellular functions.
The future has begun: AAV gene therapy in SPG50 and ASOs in KIF1A-associated disease
In 2024, Dowling et al. presented a roadmap for personalized treatment of SPG50, reporting the development of a gene replacement therapy specifically designed for a single patient with this HSP subtype. 74
SPG50 is an ultra-rare HSP caused by biallelic pathogenic variants in AP4M1, which encodes a subunit of the adaptor protein complex 4 (AP-4) involved in intracellular vesicle trafficking. 74 Mutations in AP4M1 lead to a loss of gene function and a disruption of AP-4 assembly, impairing the transport of key neuronal proteins. This study is the first-ever individualized gene therapy trial for SPG50. 74 Developed within 3 years of the patient’s diagnosis, the therapy used an AAV9-based vector (AAV9-AP4M1) delivered intrathecally. The trial prioritized safety and showed no serious adverse events or evidence of neurotoxicity over 12 months. Extensive immunosuppression was effective in preventing immune responses to the transgene. Efficacy assessments suggested potential stabilization of the disease, with improvements in motor function and developmental scores, and no observed deterioration or loss of abilities.
At the same time, Ziegler et al. reported the use of an allele-specific ASO in a nine-year-old patient with a severe form of KIF1A-associated neurological disorder (KAND), a condition that can phenotypically overlap with HSP and is caused by de novo heterozygous missense variants in KIF1A. 75 The patient carried the pathogenic p.Pro305Leu variant, known to exert a dominant-negative effect. A custom-designed 2′-O-methoxyethyl-modified gapmer ASO (nL-KIF1-001) was administered intrathecally over a 9-month period. The therapy selectively degraded the mutant transcript while preserving the wild-type allele. Aside from a transient cerebrospinal fluid complication after the first dose, the treatment was well tolerated. Clinically, the patient experienced a marked reduction in refractory spells of behavioral arrest, fewer falls, qualitative improvements in gait, increased independence, and stable cognitive function.
The first of these two cases illustrates feasibility in ultra-rare settings, though still preliminary. The second provides preliminary evidence supporting the safety and therapeutic potential of allele-specific ASOs for dominant-negative forms of neurodegenerative disease, such as KAND, and by extension, opens avenues for tailored molecular therapies in selected cases of complex HSP.
Limitations and future challenges of gene therapies
Despite the revolutionary potential of gene therapy and gene editing approaches, several significant challenges remain for their application in HSP. One major limitation is the economic burden associated with the development of highly specialized therapies for rare diseases, which often fail to attract substantial investment from the pharmaceutical industry. Furthermore, the considerable genetic heterogeneity of HSP implies that, in principle, each pathogenic variant might require a distinct therapeutic strategy, hindering scalability and broad application. However, emerging research suggests that identifying shared pathogenic pathways across different HSP subtypes—and possibly across related disorders like spinocerebellar ataxia (SCA), leukodystrophy, PLS, and others—could allow the development of common therapeutic targets, overcome the rarity barrier, and make gene therapy more feasible and economically sustainable in this context.
In addition to these economic and scientific challenges, the long-term safety and feasibility of gene therapy in HSP must also be carefully considered. Many HSP subtypes follow a very slow progressive course, meaning that the acceptable risk threshold for irreversible interventions is lower than in rapidly disabling conditions. A major concern is the potential for delayed adverse events, including insertional mutagenesis, off-target genome editing, persistent vector latency, and immune-related toxicity, which may only emerge years after treatment.76,77 Regulatory agencies such as the FDA and European Medicines Agency (EMA) therefore recommend extended follow-up of at least 5 years for AAV vectors and up to 15 years for integrating or genome-editing approaches, supported by standardized monitoring protocols. 77 Up-front toxicity associated with conditioning regimens in ex vivo strategies also raises particular concerns in disorders with slow progression; antibody-based conditioning strategies are under development to mitigate these risks. 78
Another key limitation is the difficulty of redosing: while AAV vectors ensure durable expression, immune responses often prevent repeated administrations. Alternative DNA-based platforms that are re-dosable, as well as inducible or on-demand gene expression systems, are emerging to provide more flexible and controlled exposure over the long course of disease.79,80 Finally, operational barriers such as patient retention, missing data, and the transition from pediatric to adult care complicate the long-term monitoring required in rare diseases. Leveraging registries and real-world data is increasingly recognized as a solution to sustain longitudinal safety and efficacy evaluation. 77 Patient engagement strategies, mobile health tools, and integration of patient-reported outcomes are also essential to ensure adherence and improve data completeness. Collectively, these economic, scientific, and operational considerations highlight both the promise and the limitations of gene therapy in HSP, underscoring the need for innovative approaches that balance risk and benefit in the context of slowly progressive neurodegenerative disorders.
Drug repurposing: a cost-effective gene therapy solution
Drug repurposing is emerging as an innovative strategy for addressing rare diseases such as HSP, offering a promising way to develop effective treatments less expensively and in significantly shorter timeframes compared with new drug development. 81 It exploits drugs already approved for other clinical conditions, bypassing the need to develop completely new drugs, which is a long, complex, and costly process (Table 3). Rare diseases, affecting only a small percentage of the population, often do not attract the interest of large pharmaceutical companies due to the limited profits to be made. In this setting, drug repurposing becomes crucial, as it helps overcome these economic challenges. Approved drugs already have established safety profiles, reducing risks during clinical trials and enabling a quicker return on investment due to the possibility of faster market access. Additionally, modern technologies such as AI and bioinformatics can now help to efficiently identify new uses for them. Computational innovations, allowing the analysis of vast amounts of genomic, epigenetic, and transcriptomic data, and advanced tools like the Connectivity Map and the Library of Integrated Network-Based Cellular Signatures, enabling the mapping of changes in gene expression profiles, can make for a more precise selection of potential drugs for repurposing. 82 In the context of HSP, this could expedite the discovery of effective therapies tailored to the genetic causes of these rare and debilitating conditions, providing a quicker, more affordable route to treatment development and giving hope to patients. 83
Comparing gene therapy and drug repurposing: benefits, challenges, and differences.

AAV, adeno-associated virus; HSP, hereditary spastic paraplegia.
The case of menatetrenone
Among emerging therapeutic strategies for HSP, the recent use of the compound menatetrenone (MK4, a clinically approved form of vitamin K2) in a single case of infantile-onset ascending hereditary spastic paralysis (IAHSP) caused by a missense variant in ALS2 illustrates the practical application of drug repurposing in HSP. 83
The study focused on a patient carrying the missense mutation R1611W in the VPS9 domain of Alsin, critical for the functional oligomerization of the protein. Using a drug repurposing approach, the authors performed virtual screening of approved compounds and identified MK4 as a candidate capable of masking the mutant residue and restoring proper Alsin tetramer formation. However, MK4 was effective only for the R1611W mutation, and multiple other ALS2 missense variants are known, in addition to many truncating mutations. As highlighted by the authors, similar studies need to be carried out for each mutation, with no guarantee of identifying a suitable compound in every case. Therefore, while the IAHSP experience provides a valuable proof-of-principle for precision approaches, its generalizability to the broader spectrum of ALS2-related disorders remains limited.
The future—targeting the patient: the rise of personalized therapy
Decoding genetic complexity: rare variants and polymorphisms as keys to precision treatment
Rare and private genetic variants account for the vast majority of pharmacogenetic variation across populations and can significantly influence individual responses to treatment. 84 Although these variants are often overlooked in genome-wide association studies and candidate gene studies, they may account for some of the “missing heritability” and explain unexpected drug responses.85,86 Their functional impact is increasingly assessed through computational prediction tools, yet clinical validation remains challenging due to the need for large cohorts. Moreover, substrate-dependent effects of certain variants, especially in pharmacogenes such as CYP2D6 and drug transporters, further complicate their interpretation.
In HSP, where genetic heterogeneity is substantial, investigating rare variants may be key to refining therapeutic strategies. As we move toward highly personalized treatments—including gene therapy—understanding the role of rare variants could help in predicting efficacy, avoiding adverse reactions, and guiding patient-specific interventions.87,88 This applies particularly to the design of genotype-tailored approaches that consider not only the primary mutation but also modifier genes and individual pharmacogenomic profiles. 89
The use of polygenic scores holds significant promise for predicting treatment response in HSP. This predictive capability is valuable not only for optimizing currently available symptomatic therapies but also as a critical tool complementing emerging gene therapies.90–92 In the complex genetic landscape of HSP, which involves multiple pathways (axonal transport, mitochondrial function, lysosomal and exosomal activity, myelination, and other cellular processes), polygenic scores can provide insight into the individual’s molecular profile. Greater understanding of the burden of variants affecting the different pathways means more precise prediction of therapeutic efficacy and better personalization of treatment strategies. Furthermore, polygenic scoring can identify underlying defects that might benefit from additional or combined gene-based interventions, opening new avenues for multi-targeted gene therapy approaches in HSP management. Thus, integrating polygenic risk assessment into clinical practice could enhance treatment precision and efficacy (Table 4).
Strategies supporting personalized therapy in hereditary spastic paraplegia.

CRISPR, clustered regularly interspaced short palindromic repeats; CYP2D6: cytochrome P450 family 2 subfamily D member 6; iPSCs, induced pluripotent stem cells.
Induced pluripotent stem cells in precision medicine and their potential in hereditary spastic paraplegia
Induced pluripotent stem cells have become a cornerstone of precision medicine, enabling the creation of patient-specific cellular models that capture unique genetic and molecular characteristics. 93 These models allow in-depth investigation of disease mechanisms, screening of therapeutic compounds, and prediction of individual responses to treatment, all within a personalized framework. iPSCs can be differentiated into various cell types, including neurons, making them especially valuable for modeling neurological diseases. One of the major advantages of human-based iPSC models lies in their ability to capture features of the individual’s genetic background, including single-nucleotide polymorphisms and rare genetic variants, which can profoundly influence drug responses. This is especially relevant in HSP, where variability in clinical presentation and progression could be linked to the patient’s unique genetic makeup. Patient-derived iPSCs allow researchers to investigate how these genetic differences impact neuronal function and treatment efficacy in a controlled in vitro environment.
When combined with gene editing technologies such as CRISPR-Cas9, iPSCs also provide a powerful platform for correcting pathogenic variants or introducing specific mutations to study their functional consequences.70,71
In the context of HSP, iPSC-derived neuronal models offer a unique opportunity to explore how different genetic variants impact neuronal development, axonal integrity, and cellular stress responses. Furthermore, they may enable the evaluation of gene therapy approaches and pharmacological treatments tailored to the patient’s individual genotype. Gene therapy approaches, including CRISPR-Cas9-mediated editing and AAV-mediated gene replacement, can be optimized in vitro using iPSC-derived neurons to evaluate efficacy, specificity, and potential off-target effects in a controlled, patient-specific setting. 94 Furthermore, by integrating transcriptomic and proteomic profiling, iPSC-based platforms can help stratify patients by molecular signatures, predict individual treatment responses, and guide the selection of gene therapy candidates.
Despite these advances, a notable limitation of iPSC-derived neurons is their immaturity compared with adult neurons, which may affect the translational validity of experimental findings. 95 However, important efforts are being made to enhance maturation through physical stimulation techniques, biochemical manipulation (genetic and metabolic), and the development of multicellular organoid platforms. The aim is to more closely replicate the complex neuronal environment and functionality observed in vivo, improving the predictive power of iPSC-based drug screening and therapeutic assessment. Moreover, integrating patient-specific factors—such as environmental influences, sex, and ethnic background—into iPSC models will further enhance their utility in precision medicine.
Looking ahead, the convergence of iPSC technology with high-throughput genomics, transcriptomics, and AI-driven data integration will accelerate the development of models to predict treatment response and disease progression. This integrated platform will empower clinicians to design bespoke therapeutic regimens that consider the patient’s unique genetic landscape and dynamic cellular phenotypes, thus heralding a new era of precision medicine for HSP.
From cure to care: redefining therapeutic priorities in HSP
There is a gradual shift in priorities in the development of medical therapies—away from the traditional treatment-oriented model to a care-centered framework that aligns with patients’ priorities in their daily lives. The FDA’s Patient-Focused Drug Development guidance underscores the importance of integrating clinical outcome assessments (COAs) that reflect how patients feel and function and what they hope to achieve, rather than relying solely on biomarker-based or clinical measures. This perspective is particularly important in neurological diseases, where symptoms are often chronic, multifaceted, and highly variable across individuals. In HSP, patients may prioritize regaining verbal communication, bladder control, or hand dexterity over marginal gains in walking endurance. These preferences reflect the subjective experience of the disease burden and emphasize the need to design interventions that are not only genetically precise but also meaningful to patients.
Current advances in gene therapy, while promising in terms of targeting the biological basis of HSP, must be complemented by such COAs, based on endpoints (patient-reported, observer-reported, or performance outcome measures) that are tailored not just to the disease mechanism, but also to patients’ own definitions of therapeutic benefit. According to the concept of “fit-for-purpose” COAs, the choice of therapeutic endpoint must reflect the patient’s situation, including physical, emotional, and social dimensions of health. This patient-centered approach allows the construction of personalized endpoints, such as those based on the patient’s “most bothersome symptom,” thereby opening the way for flexible therapeutic strategies that can be adapted to each individual’s most pressing needs.
AI and digital health tools offer unprecedented opportunities to support this paradigm. They can assist in the continuous monitoring of patient-reported data, enable adaptive trials based on real-world evidence, and help clinicians understand subtle patterns of disease progression or improvement perceived as important by the patient. Ultimately, effective care for individuals with HSP and similar conditions will require integration of molecular precision and human-centered insight, leveraging technology not just to measure outcomes, but also to listen to patients and respond meaningfully to them.
Bridging today and tomorrow: the growing impact of AI and technology in HSP care
In recent years, technological innovation and AI have revolutionized biomedicine, especially the field of rare genetic disorders. Advances in machine learning, data integration, wearable sensors, and computational biology have accelerated diagnostic processes and will improve the future therapeutic decision-making process.
The following section briefly discusses technology in disease diagnosis, clinical phenotype definition, variant interpretation, and the integration of omics data for improved pathophysiological understanding—a process that ultimately leads to patient-personalized therapeutic pathways (Figure 2).
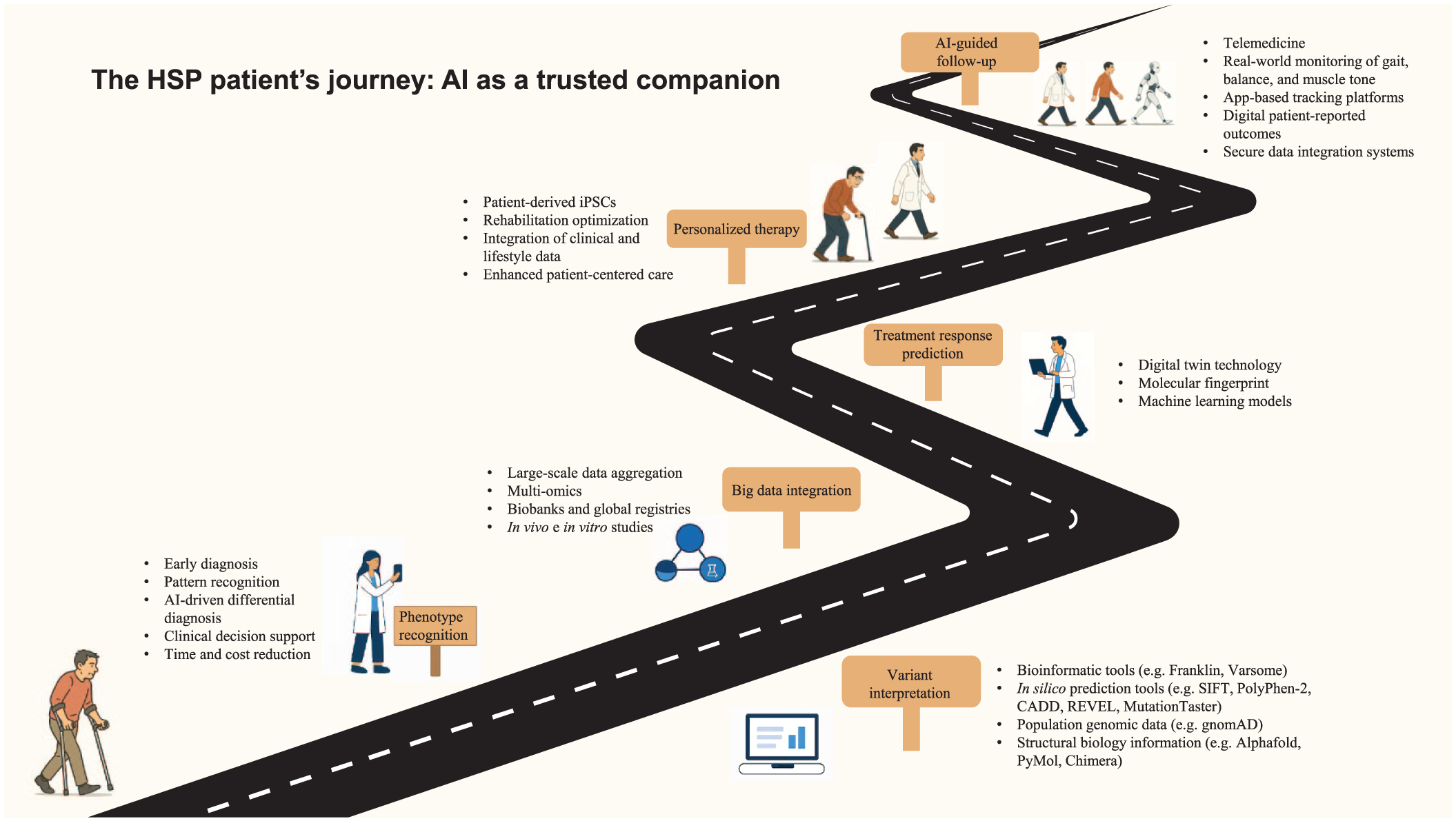
The HSP patient’s journey: AI as a trusted companion. The figure illustrates how AI, in its various applications, supports patients with hereditary spastic paraplegia throughout their disease journey.
The role of technology in the diagnosis and treatment of HSP
Modern technology is transforming the HSP diagnostic landscape. High-resolution gait analysis systems and wearable motion sensors, able to capture even subtle signs of progression96–98 and support longitudinal monitoring,99–102 are now enabling clinicians to objectively quantify motor dysfunction and stratify disease severity. Several recent studies have also provided concrete evidence in HSP cohorts. Loris et al. 103 reported in a longitudinal cohort of 55 patients that mobile digital gait analysis with inertial sensors can track disease progression over periods of up to 77 months, with gait parameters correlating with SPRS progression, fear of falling, and quality of life. Beichert et al., 104 within the multicenter PROSPAX consortium, assessed digital-motor outcomes in 65 patients with SPG7 across seven centers and found that spatiotemporal gait variability measures distinguished patients from controls, even in early stages, and correlated with mobility and activities of daily living. To reduce technical complexity, van Gelder et al. 105 evaluated a single lower-back sensor for step detection in HSP, showing high accuracy and reliability even in patients using walking aids, thereby facilitating potential large-scale application. Finally, a systematic review of 38 instrumented gait studies in HSP identified candidate digital endpoints—such as walking speed and knee range of motion, as well as more disease-specific measures like foot lift, foot range of motion, and gait variability—while emphasizing the need for further multicenter, longitudinal validation. 106 Building on these findings, wearable sensors and app-based platforms can extend their role beyond research settings, enabling continuous monitoring of gait, balance, and muscle tone in daily life.96,107–110 Such tools may help clinicians tailor interventions over time and improve the sensitivity of clinical trials, allowing subtle treatment effects to be detected in rare and heterogeneous conditions like HSP. Digital platforms also provide opportunities for remote physiotherapy and telerehabilitation, reducing the need for travel and supporting adherence to personalized exercise programs. Finally, combining longitudinal data with machine learning approaches may support the prediction of disease course and treatment response, as shown by a recent HSP study that used mobile gait analysis to evaluate the effects of BoNTA and applied predictive models to individual patient data. 111
In parallel, computational decision-support systems have become essential in accurately defining the genetic etiology of HSP. By inputting clinical, imaging, and electrophysiological data into algorithmic platforms, prioritized lists of candidate genes can be generated. Integrating diagnostic data across multiple modalities—from neuroimaging (e.g., MRI, PET) to neurophysiological evaluations like electroneuromyography, visual evoked potentials, and somatosensory evoked potentials—increases in diagnostic accuracy and guides targeted genetic testing and counseling, helping differentiate HSP from phenotypically overlapping disorders such as SCA, leukodystrophies, and certain mitochondrial diseases.98,112
In rare diseases like HSP, structural and logistical barriers often limit access to specialized care. Their low prevalence means that clinical expertise is typically concentrated in a small number of highly specialized centers, frequently distant from patients’ homes. This difficulty is compounded by the motor impairments characteristic of HSP—such as spasticity, gait disturbances, and progressive mobility limitations—which make travel physically demanding and financially burdensome. A promising and valuable alternative in this setting is telemedicine, which, involving the use of digital technologies (e.g., wearable sensors, app-based platforms, secure communication tools), enables remote consultations, virtual assessments, and ongoing monitoring. The telemedicine approach is proving increasingly capable of delivering care comparable to that provided in traditional clinical settings, and in the coming years, it is expected to play an increasingly central role in facilitating continuous, personalized, and accessible care for individuals living with rare and disabling conditions like HSP. Remote platforms enable neurologists and rehabilitation teams to conduct virtual assessments, review gait data collected by wearable devices, and track patient-reported outcomes via validated digital questionnaires. 113 This translates into less traveling for patients and caregivers, timelier follow-ups, and better overall quality of life. Moreover, by integrating data from home-based sensors in real time, clinicians gain a more comprehensive picture of patients’ functional status in their everyday environments—information that traditional in-clinic evaluations often fail to capture. 114
Taken as a whole, this interconnected technological ecosystem—from diagnostics to remote monitoring and personalized therapeutic guidance—represents an emerging approach in the care of individuals with HSP. It offers the potential for more proactive, individualized, and accessible management, ultimately improving outcomes while overcoming the constraints posed by the rarity of the disease and patients’ impaired mobility.
Variant interpretation: the role of in silico tools
Interpreting genetic variants remains one of the most significant challenges in rare disease diagnosis. However, the growing availability and sophistication of computational tools have improved the structure and reliability of variant classification into established categories—benign, likely benign, variants of uncertain significance (VUS), likely pathogenic, and pathogenic.115,116 In silico prediction tools, assessing the potential impact of nucleotide changes on protein structure and function, splicing mechanisms, and evolutionary conservation, play a critical role in this context. Tools such as PolyPhen-2, SIFT, MutationTaster, and CADD offer complementary insights into the deleteriousness of variants, particularly when combined with population frequency databases and curated clinical databases (e.g., gnomAD and ClinVar, respectively). Recent advances have led to the development of highly specialized splicing prediction algorithms—such as SpliceAI and MMSplice—which estimate the likelihood of a variant disrupting normal exon-intron recognition and pre-mRNA processing. Given the increasing evidence that splicing defects are an underrecognized mechanism in Mendelian disorders, greater importance is now being attached to these splicing prediction tools, particularly when variants fall within canonical splice sites or deep intronic regions. Moreover, integrative platforms and meta-predictors that combine multiple in silico tools (e.g., REVEL, MetaSVM) offer more robust and nuanced pathogenicity assessments, reducing ambiguity in VUS interpretation.
Together, these computational resources not only improve diagnostic yield but also enable more informed decisions regarding functional validation, family segregation studies, and potential reclassification of variants as new evidence emerges.
Integrating “big data” from omics, digital biobanks, and global patient registries
A critical foundation for advancing research in rare diseases such as HSP is the systematic collection and organization of clinical, genetic, and biological data through digital biobanks and national patient registries. These increasingly widespread infrastructures, by enhancing disease phenotype characterization, improving genotype–phenotype correlations, and supporting natural history studies, help to overcome the limitations associated with small patient populations. Moreover, they facilitate earlier and more accurate diagnoses, enable the identification of eligible patients for clinical trials, and guide the design of new studies. A notable example is the Italian HSP registry, established in 2024, which centralized clinical and molecular data at the national level to strengthen both clinical management and research efforts. 39
Building on these comprehensive and well-structured datasets, the next frontier involves integrating biological and other “big data” using powerful computational frameworks. 117 AI-driven tools can now process and synthesize data from different omics, including genomics, transcriptomics, proteomics, metabolomics, and epigenomics. 118 These datasets are often complemented by biological samples such as blood, cerebrospinal fluid, and skin biopsies. By harmonizing these heterogeneous data sources, advanced algorithms can identify novel biomarkers, elucidate pathogenic mechanisms, and uncover disease subtypes, thereby opening new avenues for precision medicine. 119 This system’s biology approach enhances understanding of disease networks by looking beyond single-gene mutations.
The ongoing expansion of digital biobanks and national registries not only supports the generation of high-quality big data but also serves as the essential foundation for developing and training advanced AI systems. By integrating and analyzing this wealth of information, AI tools can reveal hidden patterns, predict disease progression, and tailor therapeutic strategies to individual patients, ultimately ushering in a new era of personalized and precision care for rare neurological disorders.
Toward personalized treatment: fingerprinting and digital twin technology
Among the technological advances driving progress toward personalized treatment strategies for HSP, molecular signature analysis—based on the integration of genetic, transcriptomic, metabolic, and epigenetic profiles—offers a comprehensive view of the single patient’s disease state.120,121 This detailed profiling method enables the development of tailored therapeutic approaches that go beyond mere relief of symptoms, potentially encompassing gene-targeted therapies, metabolic modulators, and individualized rehabilitation programs (Table 5). By deeply characterizing patients, clinicians can better anticipate their disease progression, predict treatment responses, and optimize long-term care plans.
Technological tools in hereditary spastic paraplegia and benefits for management.

AI, artificial intelligence; CSF, cerebrospinal fluid; HSP, hereditary spastic paraplegia; SCA, spinocerebellar ataxia; VUS, variant of uncertain significance.
Digital twin technology is another innovative frontier in precision medicine, with significant potential for rare disorders.122,123 A digital twin is a virtual model of an individual patient, built from their comprehensive biological, clinical, and behavioral data. 124 This model can simulate disease progression and predict the outcomes of various treatment scenarios, offering a risk-free environment for testing therapeutic strategies before applying them in real life. In HSP, digital twins could incorporate data from genomics, imaging, gait analysis, and biomarker profiles to explore how a particular therapy—such as a neuroprotective agent or a rehabilitation protocol—might impact spasticity, neurodegeneration, or mobility over time. This approach could drastically reduce trial-and-error in clinical care and accelerate the path to personalized treatment.
Conclusion
For decades, the clinical management of HSP has remained anchored in a predominantly symptomatic approach, focused on mitigating motor disability, managing spasticity, and preserving functional independence. The rarity of these disorders and their marked genetic heterogeneity have posed major obstacles to the development of disease-modifying treatments, leaving clinicians and patients with limited options. In recent years, however, notable advances in molecular biology, gene therapy, and digital health technologies have begun to change the outlook for HSP.
Early gene-based interventions—such as the AAV9-mediated gene replacement trial in SPG50 or allele-specific ASOs in KAND—illustrate that targeted therapies for ultra-rare subtypes are technically feasible, though still at an experimental stage. These examples point to the gradual emergence of precision approaches that may, in the future, complement symptomatic care in selected forms of HSP.
Simultaneously, advances in AI and digital health technologies are beginning to influence several aspects of HSP care. From early diagnosis to mechanistic studies and therapeutic research, computational tools are increasingly integrated into clinical and scientific workflows. High-resolution motion analysis and wearable devices can provide objective measures of disease progression, while machine learning algorithms may support diagnostic accuracy by integrating multimodal clinical, genetic, and neurophysiological data. In silico platforms for variant interpretation also contribute to more reliable identification of pathogenic mutations, even in complex or ambiguous cases.
The integration, through systems biology approaches, of big data from different omics (genomics, transcriptomics, proteomics, epigenetics, and metabolomics) can offer insights into the underlying pathophysiological mechanisms of HSP. These multi-omic studies are pointing toward convergent pathways that could inform the development of shared therapeutic strategies. AI-assisted drug repurposing is an additional avenue under exploration, with early examples such as menatetrenone in ALS2-related HSP illustrating the potential of this approach, albeit with limited clinical evidence.
Global patient registries and digital biobanks are also playing a pivotal role in addressing the challenge of small patient numbers by aggregating clinical and genetic data across international cohorts. These infrastructures support natural history studies, refine genotype–phenotype correlations, and facilitate recruitment for clinical trials. Telemedicine, remote monitoring, and digital twins are further extending the reach of personalized care, allowing real-time tracking of disease progression and, in the future, the possibility of simulating treatment scenarios in silico.
In conclusion, although the therapeutic landscape of HSP continues to be dominated by symptomatic management, recent progress in gene therapy, technological innovation, and AI is laying the foundations for future targeted approaches. Most of these strategies remain in early or experimental stages, and their clinical translation will require careful validation and long-term follow-up. While effective disease-modifying treatments are not yet a reality for the majority of patients, the advances achieved so far indicate a gradual shift toward more personalized and data-driven care, providing cautious but realistic grounds for optimism.
Supplemental Material
sj-docx-1-tan-10.1177_17562864251406589 – Supplemental material for Hereditary spastic paraplegia: from decades of therapy to future innovations
Supplemental material, sj-docx-1-tan-10.1177_17562864251406589 for Hereditary spastic paraplegia: from decades of therapy to future innovations by Lorenzo Cipriano, Corrado Angelini and Filippo Maria Santorelli in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
We thank Dr. Catherine J. Wrenn for her expert editorial assistance. AI-based tools were employed for generating some graphical elements that were subsequently edited and composed by the authors.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.