
Abstract
Background:
Paroxysmal kinesigenic dyskinesia (PKD) is a rare neurological disorder, characterized by attacks of involuntary movements triggered by sudden action. Variants in proline-rich transmembrane protein 2 (PRRT2) are the most common genetic cause of PKD.
Objective:
The objective was to investigate the clinical and genetic characteristics of PKD and to establish genotype–phenotype correlations.
Methods:
We enrolled 219 PKD patients, documented their clinical information and performed PRRT2 screening using Sanger sequencing. Whole exome sequencing was performed on 49 PKD probands without PRRT2 variants. Genotype–phenotype correlation analyses were conducted on the probands.
Results:
Among 219 PKD patients (99 cases from 39 families and 120 sporadic cases), 16 PRRT2 variants were identified. Nine variants (c.879+4A>G, c.879+5G>A, c.856G>A, c.955G>T, c.884G>C, c.649C>T, c.649dupC, c.649delC and c.696_697delCA) were previously known, while seven were novel (c.367_403del, c.347_348delAA, c.835C>T, c.116dupC, c.837_838insC, c.916_937del and c.902G>A). The mean interval from onset to diagnosis was 7.94 years. Compared to patients without PRRT2 variants, patients with the variants were more likely to have a positive family history, an earlier age of onset and a higher prevalence of falls during pre-treatment attacks (27.14% versus 8.99%, respectively). Patients with truncated PRRT2 variants tend to have bilateral attacks. We identified two transmembrane protein 151A (TMEM151A) variants including a novel variant (c.368G>C) and a reported variant (c.203C>T) in two PRRT2-negative probands with PKD.
Conclusion:
These findings provide insights on the clinical characteristics, diagnostic timeline and treatment response of PKD patients. PKD patients with truncated PRRT2 variants may tend to have more severe paroxysmal symptoms. This study expands the spectrum of PRRT2 and TMEM151A variants. Carbamazepine and oxcarbazepine are both used as a first-line treatment choice for PKD patients.
Introduction
Paroxysmal kinesigenic dyskinesia (PKD) is the most common type of paroxysmal disorder characterized by recurrent and brief dystonic or choreoathetoid attacks triggered by sudden voluntary action. 1 Occurring without loss of consciousness, these attacks often manifest during childhood or adolescence. Carbamazepine (CBZ) is commonly used as a first-line treatment for PKD.
The proline-rich transmembrane protein 2 (PRRT2) gene, located on chromosome 16, was initially identified as the causative gene of PKD in 2011. It encodes a small protein rich in proline residues which is embedded in the cell membrane. The exact function of PRRT2 remains incompletely elucidated. The reported frequency of PRRT2 variants among PKD patients varies between populations, ranging from 22% to 65%, and among the identified variants, a variant known as c.649dupC is the most commonly observed.2–5 PRRT2 variants are also associated with a broad spectrum of other neurological disorders, such as infantile convulsions (IC), recently reported hemiplegic migraine and seizures, 6 which can sometimes coexist with PKD. In addition, other genes such as transmembrane protein 151A (TMEM151A), paroxysmal nonkinesigenic dyskinesia protein (PNKD), potassium calcium-activated channel subfamily M alpha 1 (KCNMA1), potassium voltage-gated channel subfamily A member 1 (KCNA1), solute carrier family 2 member 1 (SLC2A1), have been reported to be associated with PRRT2-negative patients with PKD.7–10 Collectively, the above findings highlight the clinical and genetic heterogeneity of PKD. 11
Several studies have documented clinicogenetic correlations in patients with PKD, indicating that patients carrying PRRT2 variants tend to experience their first attack at an earlier age, have longer duration of attacks and a more complex form of PKD compared to those without PRRT2 variants.3,4,6,12–16 Nevertheless, considering the clinical and genetic heterogeneity, we further summarized the clinical and genetic features of 219 patients with PKD and performed clinical–genetic phenotype analysis between the probands with and without PRRT2 variants. Given that PRRT2 variants are absent in approximately half of primary PKD patients, investigating other potential causative genes in PRRT2-negative patients would be valuable. Hence, we further applied whole exome sequencing (WES) for 49 available PKD probands without PRRT2 variants.
Methods
Standard protocol approvals, registrations and patient consent
A total of 219 patients diagnosed with PKD were enrolled in our study from 2014 to 2022. To exclude secondary factors, routine neuroimaging, electrolytes analysis, ceruloplasmin testing and thyroid and parathyroid hormone testing were performed in all probands diagnosed with PKD. The clinical data of all patients were obtained with a standard protocol. According to Bruno’s criteria, 1 two special neurologists determined the diagnosis after screening the clinical and genetic data.
Genotyping
Genomic DNA was extracted from peripheral blood cells sourced from the enrolled patients and their available family members. To identify PRRT2 (NM_145239.2) variants, Sanger sequencing was performed. Sanger sequencing data were then analysed and checked using Mutation Surveyor v5.1.1 software (Soft Genetics LLC., USA). WES was performed on 49 available PRRT2-negative PKD probands. The whole exome was captured using the Agilent SureSelect Human All Exon V6 Kit (Agilent Technologies, CA, USA ) and sequenced using the Illumina NovaSeq 6000 platform. Sequencing reads were aligned to human genome assembly hg38 using Burrows Wheeler Aligner. Variants were called using the Genome Analysis Toolkit HaplotypeCaller software (Broad Institute, USA) and annotated with ANNOVAR. The variants were excluded by the following criteria: (a) the variants did not affect the amino acid; (b) allele frequency >1% according to 1000genome, genomAD, ESP6500 and ExAC database. We searched various online databases including GeneCards, Orphanet, JuniorDoc online database, ClinVar, OMIM and PubMed to focus on genes known to be pathogenically mutated in paroxysmal dyskinesias (PxD). The genes are listed in Supplemental Table 1.
The pathogenicity of all variants was predicted using the following in silico tools: Mutation Taster (http://www.mutationtaster.org), Polymorphism Phenotyping v2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/), Functional Analysis through Hidden Markov Models (FATHMM-MKL, http://fathmm.biocompute.org.uk/) and Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu/). The pathogenicity assessment of the filtered variants was conducted in accordance with the American College of Medical Genetics and Genomics (ACMG) standards and guidelines using the Varsome tool (https://varsome.com/).17,18 The four missense variants including c.412C>G, c.623C>A, c.640G>C, c.439G>C and c.224C>T were predicted to be ‘benign’ or ‘likely benign’ or as polymorphisms. These variants are most likely non-pathogenic and were not included in the PRRT2-positive group. The patients with PRRT2 variants of uncertain significance were classified into the PRRT2-positive group. The topological structure of the PRRT2 protein in this study cited the work from Rossi et al. 19
Clinical and genetic analysis
The genotype–phenotype correlation was analyzed between 159 probands with and without PRRT2 variants. The clinical phenotype included sex, age of onset, IC history, family history of PKD, time to diagnosis, seizures, migraine, aura, type, frequency and duration of the attack, face involvement, laterality, falling and response to treatment. In our series, the response evaluation to anticonvulsant treatment was classified into three levels: complete (attacks vanished, with or without premonitory sensation), incomplete (occasional attacks at a low frequency) and nonresponsive/insensitive [attacks showed a decrease of <25% versus the previous level when the dosage of oxcarbazepine (OXC) or CBZ was increased to 400 or 450 mg daily], which was slightly modified based on the standard. 13 We also conducted a comparison between patients with truncated variants and those with non-truncated variants. We classified missense variants (c.856G>A, c.955G>T, c.884G>C, c.835C>T and c.902G>A) as non-truncated variants. While the variants located at the splice site (c.879+4A>G and c.879+5G>A), frameshift variants (c.649dupC, c.649delC, c.696_697delCA, c.367_403del, c.347_348delAA, c.116dupC, c.837_838insC and c.916_937del) and a nonsense variant (c.649C>T) were classified as truncated variants.
The difference in categorical variables between the two groups was assessed by Chi-square analysis. Fisher’s exact values were calculated when expected frequencies in any of the cells were below 5. Nonparametric data, continuous data and ranked data with nonnormal distributions between the two groups were analyzed by the Wilcoxon rank-sum test. All statistical analyses and graphical representations were performed using R programming language (version 3.6.1; R Foundation for Statistical Computing, Vienna, Austria). A two-sided p value of <0.05 was considered statistically significant.
Results
Clinical features
Clinical data from 219 patients with PKD, including 184 males and 35 females, are summarized in Figure 1. Among these patients, 120 cases were sporadic, while 99 were familial from 39 PKD families. The mean age of onset was 11.2 ± 3.1 years (range of 2–19 years). A total of 151 patients (75.06%) experienced aura, including abnormal sensations in the first affected limbs before the onset of attacks. The predominant triggers identified were sudden voluntary actions and emotional stress, with only a minority citing playing games or changes in temperature. The duration of attacks of dystonia (41.41%) or chorea (6.57%) alone or in combination (52.02%) lasted less than 1 minute in all patients. During the attacks, face involvement and falling were reported by 58.97% and 17.51% of patients, respectively. The mean interval from onset to diagnosis in all probands was 7.94 years. In addition, 41 patients (23.78%) with PKD had a history of IC, 12 patients (6%) had one to two generalized tonic–clonic seizures and 11 patients (6.11%) had a history of migraine.

Clinical characteristics of 219 patients with PKD in the present study.
A total of 163 patients accepted anticonvulsant treatment, including 59 patients treated with CBZ (50–200 mg daily), 89 with OXC (150–450 mg daily) and 15 with other anticonvulsants (phenytoin, lamotrigine and topiramate). Among those receiving anticonvulsant treatment, 95.71% achieved complete or incomplete remission. Notably, among patients treated with OXC or CBZ, 98% (145/148) achieved complete or incomplete remission. While among patients using other anticonvulsant drugs, 73.3% (11 of 15) achieved complete remission or incomplete remission.
PRRT2 screening
We detected 16 variants in 70 probands, including 2 variants located at the splice site (c.879+4A>G and c.879+5G>A), 5 missense variants (c.955G>T/p.V319L, c.835C>T/p.P279S, c.856G>A/p.V286M, c.902G>A/p.G301E and c.884G>C/p.R295P), 1 nonsense variant (c.649C>T/p.R217*) and 8 frameshift variants (c.649dupC/p.R217Pfs*8, c.347_348delAA/p.K116Rfs*17, c.367_403delGGGTCCAGGCTGGAGTCTGCAGCCCCACCTGAACCAG/p.G123Pfs*41, c.649delC/p.R217Efs*12, c.837_838insC/p.M280Hfs*61, c.116dupC/p.G40Rfs*94, c.916_937delGCCCAGCGTCTGGGCCGGGTAG/p.A306Pfs*4 and c.696_697delCA/p.H232Qfs*10). Among the 70 PKD patients with PRRT2 variants, 53 (75.71%) had the c.649dupC variant, which was the most frequent. The c.649delC variant was detected in three patients. Other PRRT2 variants were detected in one patient, respectively.
Seven novel variants were confirmed after comparison with the databases mentioned above (c.367_403delGGGTCCAGGCTGGAGTCTGCAGCCCCACCTGAACCAG, c.347_348delAA, c.835C>T, c.116dupC, c.837_838insC, c.916_937delGCCCAGCGTCTGGGCCGGGTAG and c.902G>A). The sequencing results of seven novel variants were shown in Figure 2. The positions of the 16 variants of the PRRT2 gene identified in our series were shown in Figure 3. The in silico pathogenicity prediction of all variants was presented in Table 2. Among the seven novel variants, we confirmed two pathogenic variants (c.367_403del and c.835C>T) and five likely pathogenic variants (c.347_348delAA, c.902G>A, c.837_838insC, c.916_937del and c.116dupC) (Tables 1 and 2).

The seven novel variants identified in PRRT2 gene.
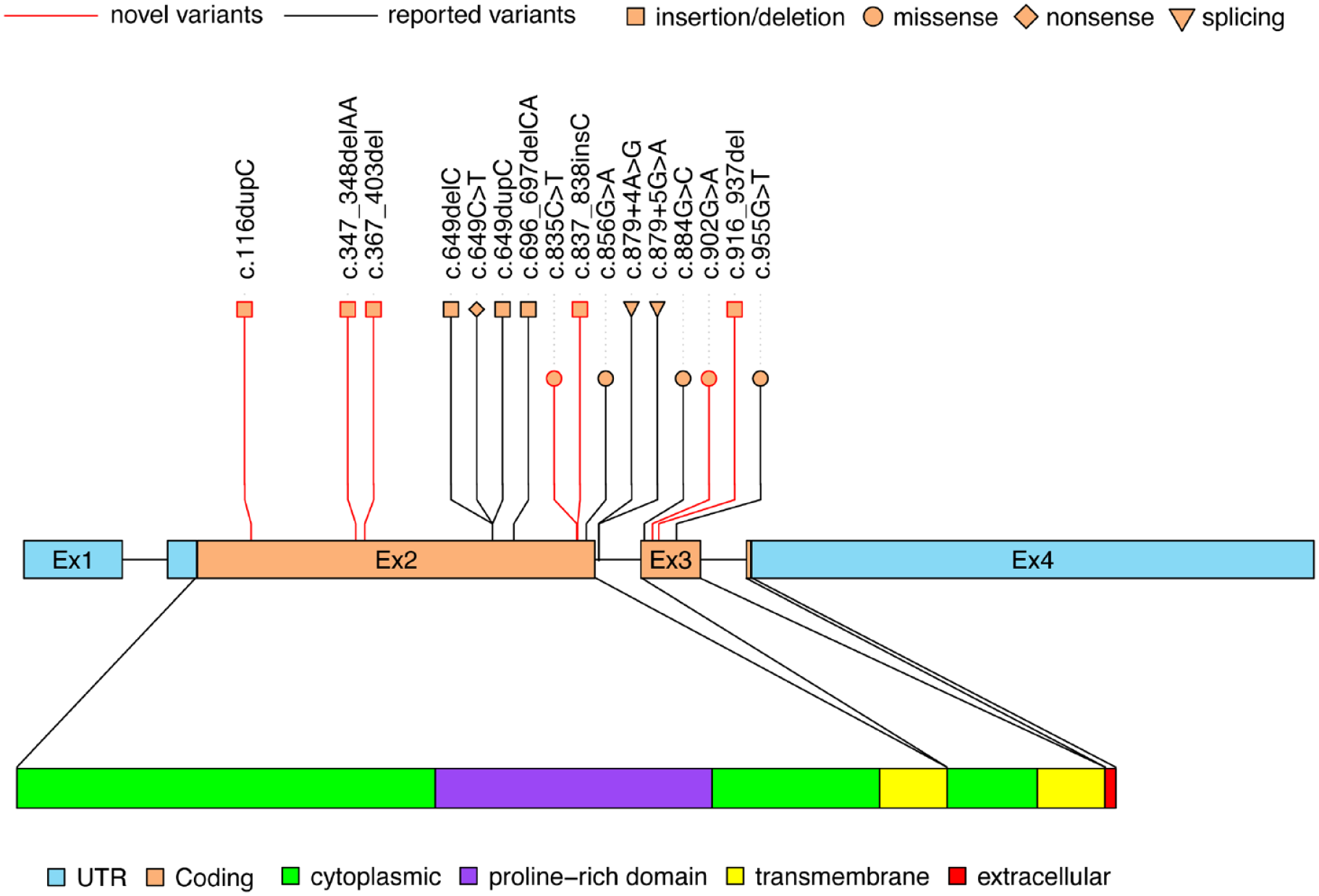
The position of the 16 variants of PRRT2 gene identified in our series are shown. The PRRT2 gene has four exons that encode a 340 amino acid protein, including two cytoplasmic regions, the first of which contains a proline-rich domain, two transmembrane regions and an extracellular region.
Genotype–phenotype correlation in 159 PKD probands with and without PRRT2 variants.

Chi-square test.
Time to diagnosis: the interval from first alert symptoms to the diagnosis of PKD in patients.
The Wilcoxon rank-sum test.
Fisher exact.
The prediction result of ACMG and in silico mutation pathogenicity in 16 variants identified in our study.

c.367_403del: GGGTCCAGGCTGGAGTCTGCAGCCCCACCTGAACCAG.
c.916_937del: GCCCAGCGTCTGGGCCGGGTAG.
BP4, supporting evidence of benign mutation, multiple computational evidence suggests no impact on the gene; CADD, combined annotation dependent depletion; FATHMM-MKL, functional analysis through hidden Markov models; N, novel; PM1, moderate evidence of pathogenic mutation, mutational hot spot; PM2, moderate evidence of pathogenic mutation, absent in population databases; PM5, moderate evidence of pathogenic mutation, novel missense change at an amino acid residue where a different pathogenic missense change has been seen before; PP3, supporting evidence of pathogenic mutation, multiple computational evidence support a deleterious effect on the gene; PP5, reputable source recently reports variant as pathogenic but the evidence is not available to the laboratory to perform an independent evaluation; PVS1, very strong evidence of pathogenic mutation, predicted nonsense mutation; R, reported.
Identification of TMEM151A gene variants
WES was performed in the 49 PRRT2-negative probands with PKD. A total of two heterozygous variants in TMEM151A were detected in two probands: a novel variant (c.368G>C), which was identified in one proband’s mother with the PKD phenotype, and a reported variant (c.203C>T), which was not detected in the patient’s parents. The sequencing results of the two variants were shown in Supplemental Figure 1. Detailed clinical information of the three patients was shown in Supplemental Table 2. The pathogenicity prediction for the two variants was presented in Supplemental Table 3. Copy number variants and other possible pathogenic genes were not detected in these cases.
Genotype–phenotype correlation
We identified a total of 159 unrelated patients with PKD, including 70 PRRT2 variant carriers and 89 non-carriers. Among these patients, PRRT2 variant carriers were more prone to have a positive family history and experience IC than non-carriers. The PRRT2 variant carriers also exhibited an earlier onset (10.19 versus 12.62 years) and were more likely to fall during attacks (27.14% versus 8.99%). Delays to diagnosis were noted, with PRRT2 variant carriers experiencing a delay of 10.69 years, compared to 5.85 years in non-carriers (p = 0.001). No significant difference was observed in the assessment of other clinical attacks or the response to anticonvulsant treatment between the two groups. A significant difference in laterality was identified between patients with truncated variants and non-truncated variants (p = 0.017) (shown in Supplemental Table 4).
Discussion
PKD is the most common type of PxD. Variants in the PRRT2 gene are the main cause of primary PKD. Here, we analyzed the genotype–phenotype correlation of 219 PKD individuals and screened for other potential causative genes in 49 PRRT2-negative probands. The findings of our study further delineate and enlarge the clinical and genetic spectrum of PKD.
Most PKD patients in our study reported the typical clinical characteristics including kinesigenic and emotion-related triggers, aura and dystonic attacks less than 1 min in duration, which further corroborates previous findings. We observed a 79% prevalence of aura in our series, which aligns with previous results showing a prevalence ranging from 48% to 82% in PKD patients, irrespective of PRRT2 variant status.3,4,6,14,15 Numbness, tingling and muscle weakness in the affected limbs, extending from the lower limbs to the upper limbs, were the most common forms of aura. The origin of aura remains undetermined, although advanced neuroimaging studies have demonstrated abnormal changes in the primary somatosensory area, which may be associated with the emergence of auras in PKD patients.20,21
In our study, OXC rather than CBZ was mostly applied, where 89 patients received OXC treatment, while 59 patients were administered CBZ. Both OXC and CBZ demonstrated remarkable effectiveness, but some patients continued to experience auras with no further attacks, indicating both CBZ and OXC can be used as the first-line drug for PKD patients. It was supported by some recent studies that revealed that PRRT2 interacts with Nav1.2/1.6 channels, causing a reduction of Na+ current, an induced negative shift in voltage-dependent inactivation and a deceleration in the recovery from inactivation.22,23 Therefore, loss of PRRT2 leads to an increase of spontaneous and evoked activity and increased excitability of neurons, which mechanistically explains the successful therapeutic control of paroxysmal attacks of PKD patients with PRRT2 variants treated by Na+ channel blockers such as CBZ or OXC. Notably, 16% of patients did not take medications either due to having less frequent attacks or out of fear of its side effects. During follow-up, we observed that some patients achieved remission with dosages of medication lower than the conventional practice (only once or twice per week), and remission appeared to be age-dependent. Given these findings, the treatment strategies should be individualized according to the natural remission history of attacks and the patient’s social and occupational requirements.3,4
PKD with IC (PKD/IC), also known as IC and choreoathetosis syndrome, typically presents as benign, afebrile IC within the first year of life, and these patients experience PKD in early childhood. In our series, we observed a higher prevalence of PKD/IC compared to the findings reported by Huang et al. 3 and Liu et al. 4 We also observed that PRRT2 variant carriers were more prone to have a history of IC than non-carriers. The link between developmental differences in PRRT2 expression and these temporal changes across different life stages remains unknown. In our series, 6.1% of PKD patients reported having migraines, which aligns with the 4.5–6.2% range reported in previous studies.3,4 A recent large-sample study revealed PRRT2 variations in 30 of 860 probands (3.5%) with suspected hemiplegic migraine, thus, PRRT2 should be regarded as the fourth autosomal dominant gene for hemiplegic migraine. 24 In addition, 6% of participants in our study experienced one to two unprovoked generalized tonic–clonic seizures during childhood or adolescence. Another study documented that 3 out of 18 familial PKD patients had concurrent generalized tonic–clonic seizures and rolandic epilepsy. 6 Additionally, several cases combined with PKD and epilepsy with CHRN4A variants have been reported. 8 Investigating the molecular mechanisms and exploring unidentified non-genetic factors may therefore be necessary to better understand the reasons behind this phenotypic overlap.
We are the first to report a higher incidence of falls during attacks before treatment in PKD patients with PRRT2 variants, suggesting that PRRT2 variant carriers experienced more severe attacks than non-carriers. Notably, the mean interval from onset to diagnosis in our series was 7.94 years. However, the interval is shorter in PKD patients without PRRT2 variants than those with PRRT2 variants, which seems counterintuitive. A plausible explanation is that PKD patients with PRRT2 variants manifest their symptoms at a younger age. The earlier onset would make it more challenging for them to recognize and detect their attacks, delaying their decision to seek medical attention. The PKD patients were frequently misdiagnosed with epilepsy, 25 neurofunctional disorders or calcium deficiency. Despite the identification of PRRT2 in PKD improved reliability and accuracy of diagnosis, this study underscores the need to shorten the interval to diagnosis, implying that clinicians need to expand their knowledge and understanding.
In this work, we identified seven novel variants including five frameshift and two missense variants, predicted as pathogenic or likely pathogenic. A previous study reported that the missense variants in PRRT2 at the C-terminus often exhibit defects in targeting the plasma membrane, 26 which may contribute to the pathogenesis of the two novel missense variants (c.835C>T and c.902G>A) located at the C-terminus of PRRT2 protein. Future functional studies are expected to assess the impact of these two variants. The c.884G>C and c.879+5G>A variants, while not previously reported in relation to the PKD phenotype, have been associated with benign familial infantile epilepsy and febrile seizure-related epilepsy. Significantly, we are the first to report that patients with truncated variants of PRRT2 tend to have bilateral attacks, while those with non-truncated variants tend to have unilateral attacks. This distinction may be attributed to truncated variants of PRRT2 inducing profound protein dysfunction, thereby resulting in more severe paroxysmal symptoms. However, a significant disparity exists in sample sizes when comparing the differences between truncated and non-truncated variants (65 versus 5). Further confirmation of this conclusion is thus required in future studies.
A relatively rare subset of patients carrying homozygous or compound heterozygous variants of PRRT2 manifest with severe phenotypes with developmental delay and intellectual disability,27,28 although these variants were not detected in our series. Overall, the ‘gene–dosage effect’ and environmental factors that influence the clinical presentations and cause severe impairments remain unidentified in PRRT2-associated disorders. It has been proposed that the variants of PRRT2 affect neurotransmitter release 29 and hyperexcitability of certain brain circuits, particularly those involving the basal ganglia and its connections with the thalamus, cortex and cerebellum. In addition, the PRRT2 protein exhibits predominant expression in these regions, which are implicated in the clinical characteristics of diseases linked to PRRT2, 30 but the function and the pathogenic mechanisms associated with it remain unclear.
However, the prevalence of PRRT2 variants in PKD patients varies from 27% to 65%, suggesting that additional causative genes are yet to be discovered.5,6,12,14,15,31 Here we detected two missense variants including a novel variant in TMEM151A in 49 PRRT2-negative PKD patients. Recently, variants in TMEM151A have been identified as causative in PKD. 10 The overall frequency of TMEM151A in our study was 4.1% (2/49), aligning with the 4.8% reported by Tian et al. 32 TMEM151A, located at 11q13.2, is a relatively newly discovered gene with functions that currently remain unknown. One study reported that patients with TMEM151A variants are more likely to be sporadic, experience shorter attacks and present with dystonia compared to those with PRRT2 variants, although these findings require further confirmation due to a relatively small sample size. 33
Limitations
This study has several limitations. First, the data presented were retrospective, which may have caused selection bias, although follow-up with these participants will continue. Second, detailed clinical information about some family members was missing. Finally, we did not provide functional verification for the new PRRT2 variants.
Conclusion
In conclusion, we expanded the variant spectrum of PRRT2 and TMEM151A genes. Genotype–phenotype analyses revealed a significant association between PRRT2 variants and an earlier age at onset, serious manifestations and attacks, delay in diagnosis and a complicated form of PKD. Furthermore, we first reported that patients with truncated variants of PRRT2 tend to have bilateral attacks, suggesting the presence of more severe paroxysmal symptoms.
Supplemental Material
sj-docx-1-tan-10.1177_17562864231224110 – Supplemental material for Genetic and phenotypic analyses of PRRT2 positive and negative paroxysmal kinesigenic dyskinesia
Supplemental material, sj-docx-1-tan-10.1177_17562864231224110 for Genetic and phenotypic analyses of PRRT2 positive and negative paroxysmal kinesigenic dyskinesia by Yingying Zhang, Jiechuan Ren, Tianhua Yang, Weixi Xiong, Linyuan Qin, Dongmei An, Fayun Hu and Dong Zhou in Therapeutic Advances in Neurological Disorders
Supplemental Material
sj-tif-2-tan-10.1177_17562864231224110 – Supplemental material for Genetic and phenotypic analyses of PRRT2 positive and negative paroxysmal kinesigenic dyskinesia
Supplemental material, sj-tif-2-tan-10.1177_17562864231224110 for Genetic and phenotypic analyses of PRRT2 positive and negative paroxysmal kinesigenic dyskinesia by Yingying Zhang, Jiechuan Ren, Tianhua Yang, Weixi Xiong, Linyuan Qin, Dongmei An, Fayun Hu and Dong Zhou in Therapeutic Advances in Neurological Disorders
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.