
Abstract
Background:
Stiff person syndrome (SPS) is a rare slowly progressive autoimmune neuronal hyperexcitability disease with very-high GAD-65 antibody titers that most commonly presents above the age of 20, with muscle stiffness, painful muscle spasms, slow gait, and falls leading to disability. In other autoimmune disorders, late-onset disease has different symptom-spectrum and outcomes, but there is no information regarding late-onset SPS (LOSPS).
Objective:
Highlight delayed diagnosis and poor tolerance or incomplete response to therapies of patients with LOSPS and outline how best to increase disease awareness early at onset.
Design: A retrospective chart review.Methods:
We reviewed GAD-positive SPS patients with symptom onset above age 60, identified among 54 SPS patients, examined, treated and followed-up by the same clinicians, focused on clinical presentation, misdiagnoses, response and tolerance to therapies, and evolved disability.
Results:
Nine patients had LOSPS with symptom onset at median age of 61 years (range 60–78), and current median age of 73. The median time from symptom onset to SPS diagnosis was 3 years; prior to diagnosis, five patients were treated for lumbosacral radiculopathies (one with laminectomy), two for Parkinson’s disease, one for multiple sclerosis, and another for cerebellar degeneration. Progressive decline occurred rapidly in all patients; at time of diagnosis, six patients were already using a cane or walker and two were wheelchair-bound. Tolerance and response to treatment were limited; two patients did not respond to IVIg, two discontinued IVIg despite early response due to comorbidities (cardiac disease, thrombosis), four others partially responded to IVIg and one to rituximab; several could not tolerate high doses of oral antispasmodics due to somnolence; and two patients died.
Conclusions:
LOSPS is almost always misdiagnosed for other similar conditions commonly seen in the elderly. Patients with LOSPS decline quickly to clinically severe disease due to delayed treatment initiation, poor response or tolerance, other comorbidities, and possibly immunosenescence. Increased awareness that SPS can occur in the elderly mimicking other disorders is important for early diagnosis and treatment, even necessitating earlier immunotherapy initiation, compared to their younger counterparts, to prevent faster-evolving severe disability.
Introduction
Stiff person syndrome (SPS) is an autoimmune disorder more common in women than men above the age of 20 years, characterized by stiffness of the axial and limb muscles, episodic painful muscle spasms triggered by anxiety, emotional upset, task-specific phobias and startles from unexpected visual, tactile and auditory stimuli, that collectively result in slow gait and uncontrolled falls.1–4 The disease is due to autoimmune neuronal hyperexcitability connected to impaired reciprocal GABAergic inhibition 1 associated with very-high Glutamic Acid Decarboxylase-65 (GAD-65) antibody titers in serum and antibody presence in the CSF.1,5,6
Although in many series the median age of onset in SPS patients is around 40 years, several patients have late-onset SPS (LOSPS) with symptom onset above the age of 60. It is important to recognize LOSPS because, based on many patients we are following, the diagnosis in this age group is often delayed, resulting in increased disease progression and significant disability due to poor tolerance of antispasmodic and immunotherapeutic agents owing to frequent comorbidities in older people. In other autoimmune disorders, like myasthenia gravis, multiple sclerosis, and now Myelin Oligondendrocyte Glycoprotein Antibody-associated Disease (MOGAD), subcategories of patients with disease onset at different ages share distinct demographics, diagnostic and immunological characteristics, prognosis, and therapeutic outcomes. These have been highlighted first in patients with myasthenia gravis and late-onset disease with symptom onset above age 50 and a very late-onset disease above 65. 7 Late-onset multiple sclerosis in patients with symptoms onset above the age of 50 has also been increasingly recognized as a subset of MS with distinct features. 8 It also became apparent recently that even MOGAD and neuromyelitis optica spectrum disorder (NMOSD) also have different phenotypes according to the age of onset with late-onset NMOSD, defined at symptom onset above the age of 50, having worse prognosis due to greater severity of symptoms and rapid disease progression even without differences in the presenting clinical phenotype. 9 Age of onset also plays a role in autoimmune encephalitis as N-methyl-D-aspartate receptor (NMDAR) encephalitis predominates in younger women whereas Leucine-rich glioma-inactivated 1 (LGI1) encephalitis in older men.
There is no information however regarding LOSPS. In our early large longitudinal study done 20 years ago with 57 patients, we personally followed for 2 years at the NIH, the median age of onset was 42 years (range 20–60). 10 At that time, the disease was poorly recognized and very often misdiagnosed. In a recent series of 173 patients, referred for SPS based on clinical presentation, symptom onset above the age 60 was not infrequent. 11 In our retrospective review of 36 patients (29 women and 7 men) treated by our group over the span of 12 years with maintenance Intravenous Immune globulin (IVIg), the median current age of patients was 58 years. 12 The cut-off limit above the age of 60 in defining LOSPS is therefore representative of a subgroup of patients outside the typical range at disease onset.
We report a series of LOSPS patients that we followed since 2011 highlighting diagnostic and therapeutic challenges pertinent for the practicing neurologists to consider the disease early, avoid misdiagnoses, and treat it in a timely manner because in the elderly patients cumulative disability carries poor prognosis.
Methods
Data on patients with typical SPS, diagnosed by the same neurologists based on the previously published diagnostic criteria, 13 followed in our clinic within the last 12 years (2011–2023) were reviewed and analyzed. Demographic information and diagnostic data, including GAD antibody positivity, age from symptom onset to diagnostic confirmation, clinical presentation, early misdiagnoses, clinical response, and tolerance to therapies they have received or currently receiving, complications, comorbidities, and outcomes with evolving disability were examined and specifically analyzed.
Results
Clinical diagnostics
We identified nine patients with LOSPS. Their median age of symptom onset was 61 years and their median current age 73 years. Median time from symptom onset to diagnosis was 3 years with frequently delayed proper treatment initiation due to initial misdiagnosis. Five patients were treated for lumbosacral radiculopathy, two for osteoarthritis, two for Parkinson’s disease, and one for multiple sclerosis (Table 1). One patient, in the late seventies, underwent laminectomy and detethering surgery for spasms attributed to worsened spina bifida occulta. At time of diagnosis, six patients were already using a cane or walker and two were wheelchair-bound.
Characteristics of patients with LOSPS.
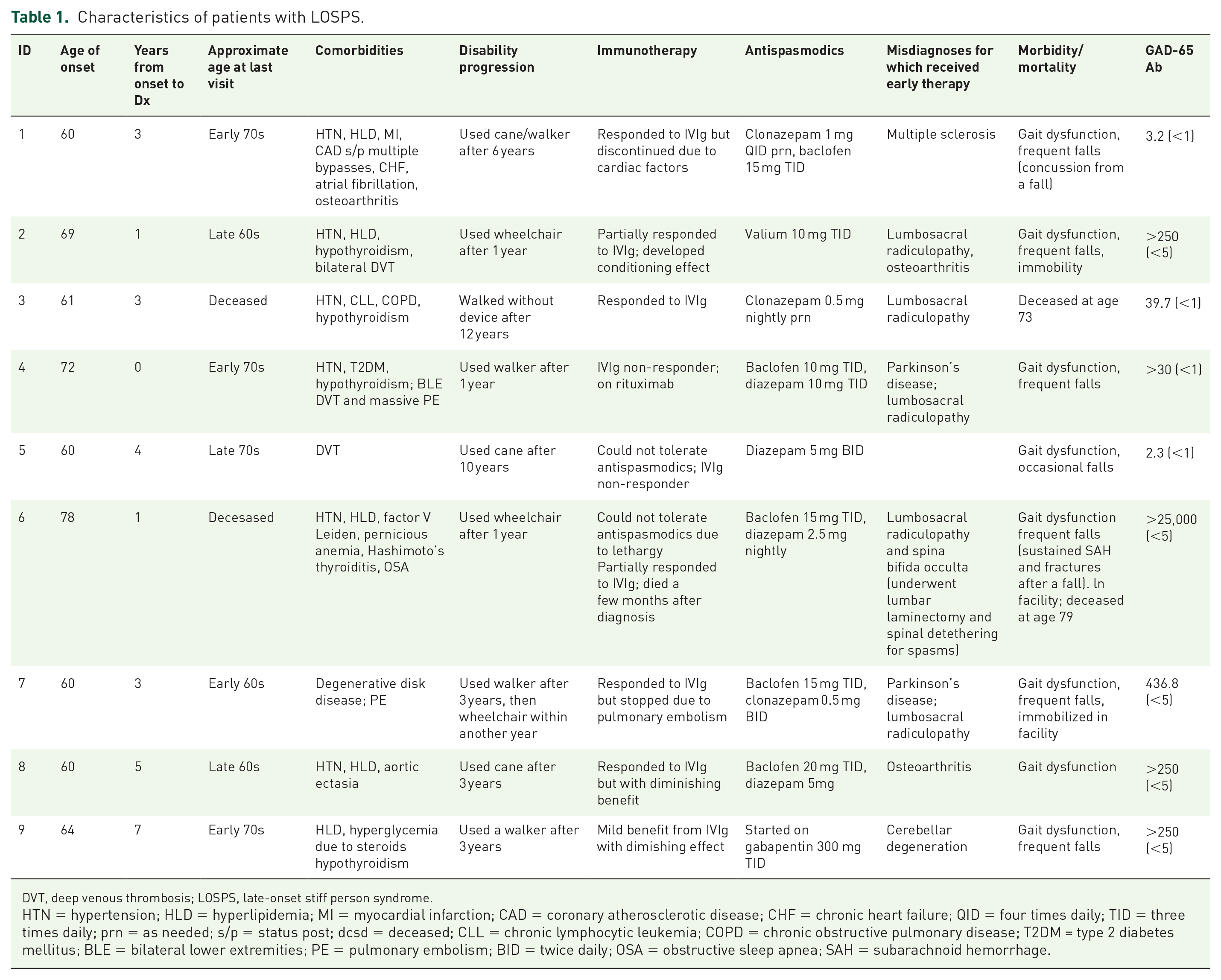
DVT, deep venous thrombosis; LOSPS, late-onset stiff person syndrome.
HTN = hypertension; HLD = hyperlipidemia; MI = myocardial infarction; CAD = coronary atherosclerotic disease; CHF = chronic heart failure; QID = four times daily; TID = three times daily; prn = as needed; s/p = status post; dcsd = deceased; CLL = chronic lymphocytic leukemia; COPD = chronic obstructive pulmonary disease; T2DM = type 2 diabetes mellitus; BLE = bilateral lower extremities; PE = pulmonary embolism; BID = twice daily; OSA = obstructive sleep apnea; SAH = subarachnoid hemorrhage.
Disease progression
Progressive clinical decline occurred relatively quickly. After a median of 3 years from symptom onset, the patients had significant gait dysfunction and frequent falls; eight out of nine patients required a device to ambulate. Two patients were residing in a care facility at the last visit due to severe level of disability. Two other patients died, one with symptom onset at the late seventies died about a 1 year later, and the other with symptom onset at the early sixties (but diagnosed 3 years later) died in the early seventies.
Comorbidities
The most frequent comorbidities were hypertension (seen in six of nine patients), thromboembolic events (seen in five patients), and hyperlipidemia (seen in five patients).
Comparison of clinical characteristics and prognosis among different age groups
No difference in the clinical phenotype was identified between younger-onset or LOSPS patients. The delayed diagnosis in LOSPS was likely related to lower clinical suspicion by the treating physicians to consider SPS as a diagnostic possibility because in this age the patients’ presenting symptoms of muscle spasms, stiffness, and fear of falling were typically viewed as being musculoskeletal in nature. As a result, the patients were most often referred to other specialties, mostly orthopedists, who focused on common for that age degenerative spine disease, interpreting their symptoms as related to cervical or thoracolumbar radiculopathies and spinal stenosis with one of them even having cervical laminectomy and fusion in the late seventies. Even when seven out of nine patients were referred to neurologists prior to coming to our clinic, there was still further delay in diagnosis because the symptoms of spastic gait and stiffness in that age were interpreted as Parkinsonism or myelopathy from spinal stenosis. Consequently, while they were undergoing other non-specific treatments, their underlying SPS was progressing while deconditioning was contributing to poor mobility.
Response to therapies
Tolerance and response to treatments were limited. All nine patients were tried on IVIg with variable response and tolerance (Table 1). Among them, two did not respond; two discontinued IVIg early, despite positive response, due to cardiovascular comorbidities and thromboembolic events or receiving anticoagulation; one started to respond but died soon thereafter ; two partially responded or showed diminishig benefit; and one other exhibited a conditioning effect, commonly seen during chronic IVIg maintenance therapy due to fear that discontinuing it will worsen their disease or lead to disease progression. 14 Regarding B-cell therapies, only one (patient #4, Table 1) received rituximab since 2018. Several patients exhibited poor tolerance to high doses of oral antispasmodics including combinations of clonazepam, diazepam, and baclofen (see Table 1), whereas two of them could not tolerate them due to somnolence.
Discussion
LOSPS with disease onset above the age of 60 is often misdiagnosed for other conditions commonly seen in the elderly populations, especially lumbosacral and cervical radiculopathies, complicated by unnecessary surgeries and other orthopedic therapies, necessitating the need for increased awareness by the practicing neurologists. Patients with LOSPS appear to have clinically more severe disease, not necessarily related to poorer response to immunotherapy, but rather due to a combination of factors, including: (a) faster disease progression related to delayed diagnosis and therapy initiation; (b) poor tolerance to antispasmodics and benzodiazepines which, in contrast to the younger SPS patients, 14 are poorly tolerated in the elderly because of excessive somnolence and falls; (c) degenerative arthritic changes in joints, cervical, and thoracolumbar spine that, even if not causative, contribute to pain and enhance symptom severity; and (d) deconditioning due to poor mobility that occurs faster in older-age patients due to cumulative effect of other comorbidities which, in combination with SPS-related stiffness, muscle spasms, and gait dysfunction, lead to early reliance on assisting devices.
The experience from the present series suggests that personalized treatment regimens are required for older patients with SPS because their poor tolerance to antispasmodics and sedating analgesics necessitates the need for earlier initiation of immunotherapy with IVIG, which is considered first-line immunotherapy. 14 Because SPS is a progressive disease, 10 treatment with IVIg should start as soon as antispasmodics are ineffective or poorly tolerated. 14 Despite their rapidly evolving disability however, our data show that LOSPS patients did not seem to have a significantly lower rate of response to IVIg because all nine patients were treated with IVIg and only two were non-responders; this represents a response rate similar to our prior study which identified a 67% responder rate to IVIg, although in LOSPS their response was partial and seemed to decline faster. 12 Rather, the limiting factor in the use of IVIg as continuing therapy in LOSPS is more related to tolerance and the existence of serious comorbidities due to cardiac problems, history of thromboembolic events, or poor venous access that necessitate early discontinuation; further subcutaneous IgG, which is preferable when IVIg is not tolerated or not considered safe, cannot be given to those LOSPS receiving anticoagulant therapies. Accordingly, in certain circumstances, rituximab which is also effective in 40% of SPS patients based on our large controlled study, 14 may be considered earlier in the immunotherapeutic algorithm in LOSPS, compared to their younger counterparts, to prevent disability. Toward this goal, the newly discussed future immunotherapies in SPS, 14 if proven effective, may be also considered earlier in LOSPS when rituximab is not effective. Another factor possssibly contributing to poor tolerance or inadequate response to immunotherapies in LOSPS may be immunosenescence and inflammaging which can weaken the innate and adaptive immunity reducing the patients’ capacity to mount an adequate immune response. Senescent immune cells may not be also helpful in adequately improving GABAergic synaptic neurotransmission or affect the repair of synaptic cell loss that normally starts above the age of 60.
The purpose of this report is to increase awareness of LOSPS to neurologists and serve as a reminder that SPS can first present in the elderly populations, but its diagnosis is more challenging and easily missed because it very often mimics common orthopedic and musculoskeletal conditions or rigid Parkinsonism, resulting in delayed therapy initiation and faster cumulative disability. On the other hand, the recent publicity on SPS has also increased awareness of this syndrome, not only amongst clinicians but also patients themselves, especially the elderly with painful back spasms, stiffness, and musculoskeletal issues who request appointments to exclude SPS.
Anecdotally, the last few months we have seen three patients above the age of 70 who have read about SPS and believed having the symptoms they have read about. A recent study from the Mayo Clinic also enhances the common SPS misdiagnoses highlighting that most of the patients referred as SPS actually had functional disorders, resulting in unnecessary and expensive therapies. 11 Another concern with IVIg therapy in the elderly is the frequent observation of a conditioning effect, seen more often with intravenous therapies. Even if IVIg is initially effective, older patients tend to exhibit earlier declining benefits and conditioning effects compared to their younger counterparts, requesting maintenance IVIg on a regular basis, even if there is no longer any significant benefit, due to their perception that it helps fatigue and pain but mostly reduces their fear that discontinuation will worsen the disease.12,14 This can lead sometimes to frustrations with their neurologists who recommend discontinuing IVIg when there are no objective signs of worsening after prolonging treatment interval, lowering the dose, or holding infusions, to justify treatment continuation causing more anxiety and depression that worsens some of their subjective symptoms. 14 In reference to depression, the reports that serotonin re-uptake inhibitors worsen the SPS symptomatology, 15 even if not observed by us, should be also taken into consideration as depression is also increased in the elderly with physical disabilities.
In summary, and in spite of the study limitations mostly related to a small number of patients with LOSPS, the data carefully dissected among the large number of SPS patients our group has been personally following, 11 clearly show that LOSPS is not only frequently missed due to low suspicion of the disease in the elderly because their complex symptomatology of painful spasms and anxiety-driven stiffness are easily attributed to other conditions, but also because of the need for multi-disciplined care to avoid rapidly evolving disability. It is essential therefore to coordinate their care with support from the family members and the other providers to properly address all their physical and psychological needs they face from the outset.