
Abstract
Despite advances in the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and other common autoimmune neuropathies (AN), still-many patients with these diseases do not respond satisfactorily to the available treatments. Repurposing of disease-modifying therapies (DMTs) from other autoimmune conditions, particularly multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSD), is a promising strategy that may accelerate the establishment of novel treatment choices for AN. This approach appears attractive due to homologies in the pathogenesis of these diseases and the extensive post-marketing experience that has been gathered from treating MS and NMOSD patients. The idea is also strengthened by a number of studies that explored the efficacy of DMTs in animal models of AN but also in some CIDP patients. We here review the available preclinical and clinical data of approved MS therapeutics in terms of their applicability to AN, especially CIDP. Promising therapeutic approaches appear to be B cell–directed and complement-targeting strategies, such as anti-CD20/anti-CD19 agents, Bruton’s tyrosine kinase inhibitors and anti-C5 agents, as they exert their effects in the periphery. This is a major advantage because, in contrast to MS, their action in the periphery is sufficient to exert significant immunomodulation.
Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a treatable chronic autoimmune disease of the peripheral nervous system (PNS) and the most common one of the chronic autoimmune neuropathies (AN). The pathogenesis of CIDP involves autoreactive T cells, autoantibodies, complement, activated macrophages mediating demyelination and secondary axonal degeneration (Figure 1). A clinical meaningful response to first-line treatments (immunoglobulin, corticosteroids, plasma exchange) is only seen in 75–80% of CIDP patients.1,2 There is consensus that more effective immunotherapies are needed, particularly for non-responders to first-line treatments or with an aggressive disease course that requires chronic immunosuppression with an improved side-effect profile.

Pathogenesis of chronic inflammatory demyelinating polyneuropathy (CIDP) and potential targets of disease-modifying treatments (DMTs).
CIDP is rare and drugs specifically designed for CIDP are not available, which also applies to a greater extent to other AN such as multifocal motor neuropathy (MMN) and anti-myelin-associated glycoprotein (MAG) neuropathy. Thus, the prevailing strategy for drug development in AN has been to examine drugs already approved for other autoimmune conditions. A particular ‘mine’ for this repurposing strategy are disease modifying treatments (DMTs), developed and licensed for multiple sclerosis (MS), 3 or neuromyelitis optica spectrum disorders (NMOSD). The rationale is that most AN, especially CIDP, share some immunopathological features with MS, such as macrophage-mediated demyelination, and with NMOSD, such as complement activation and autoantibodies, while the availability of animal models shares methodological similarities with both of them (Table 1). 4
Overview of preclinical animal models of MS and NMOSD with their induction and histopathological hallmarks and of their ‘counterparts’ in the PNS, Guillain–Barré syndrome (GBS) and CIDP.

Abbreviations: AQP4, aquaporin-4; AT, adoptive transfer; BBB, blood–brain barrier; BNB, blood–nerve barrier; CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; EAN, experimental autoimmune neuritis; GBS, Guillain–Barré syndrome; GFAP, glial fibrillary acidic protein; IgG, immunoglobulin G; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorders; NOD, non-obese diabetic; PLP, myelin proteolipid protein; RRMS, relapsing-remitting multiple sclerosis; SAP, spontaneous autoimmune neuritis.
DMTs for MS and NMOSD as related to AN
Over the past two decades, various DMTs have been approved and licensed for MS and NMOSD. They differ in terms of efficacy (i.e. reduction of relapse rate), mechanism of action, route of administration and potential side effects (Table 2).
Indications, application routes and the principal mode of action of disease-modifying drugs currently used or evaluated in MS or NMOSD.
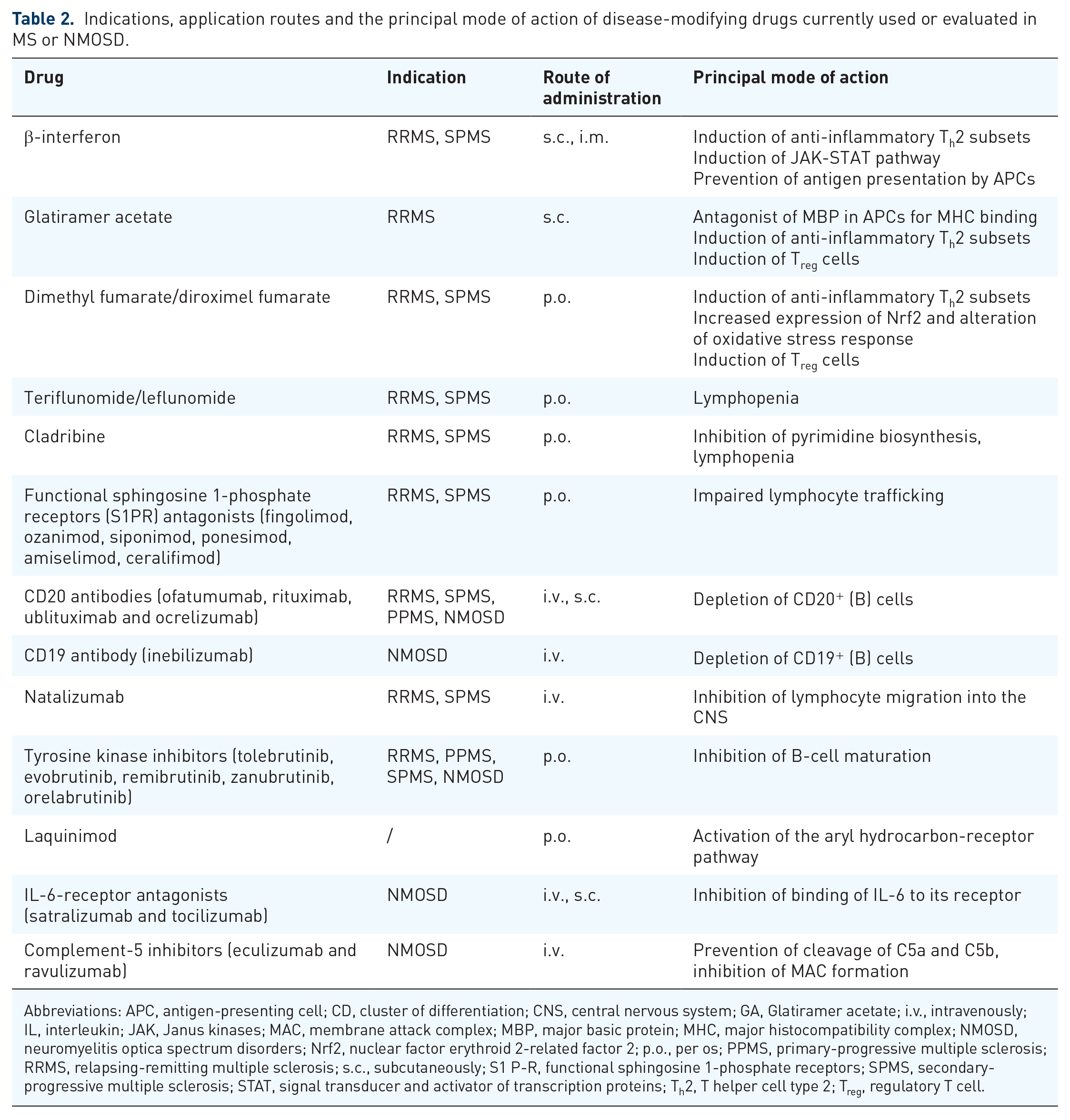
Abbreviations: APC, antigen-presenting cell; CD, cluster of differentiation; CNS, central nervous system; GA, Glatiramer acetate; i.v., intravenously; IL, interleukin; JAK, Janus kinases; MAC, membrane attack complex; MBP, major basic protein; MHC, major histocompatibility complex; NMOSD, neuromyelitis optica spectrum disorders; Nrf2, nuclear factor erythroid 2-related factor 2; p.o., per os; PPMS, primary-progressive multiple sclerosis; RRMS, relapsing-remitting multiple sclerosis; s.c., subcutaneously; S1 P-R, functional sphingosine 1-phosphate receptors; SPMS, secondary-progressive multiple sclerosis; STAT, signal transducer and activator of transcription proteins; Th2, T helper cell type 2; Treg, regulatory T cell.
Some of these have already been explored in AN, while others have been tested in preclinical models or applied to individual patients or smaller patient cohorts.
Methods: For this narrative review, we reviewed data from trials, reviews and other articles of DMTs approved for MS or NMOSD in terms of clinical mechanism of action, side effects (with a focus on PNS neurotoxicity), and available data derived from neuritis models or from use in patients with chronic autoimmune neuritis from the database PubMed. We further assessed the database for clinical trials of the U.S. National Library of Medicine between July 2021 and September 2022 for any ongoing trials for MS/NMOSD and AN. Phase I trials were excluded in the review. We excluded classical chemotherapeutics like cyclophosphamide or mitoxantrone due to their declining relevance for MS treatment algorithms and other immunomodulatory therapies like bone-marrow transplantation.
DMTs approved for moderate-active MS
Interferon-beta (IFN-β)
IFN-β is a cytokine secreted by various cells and its immunomodulatory effects are mediated through genetic activation and the Janus Kinases and signal transducer and activator of transcription proteins (JAK-STAT) signalling pathway, which is also involved in CIDP disease activity.7–10 This pathway reduces myeloid dendritic cells in the peripheral blood and prevents antigen presentation by antigen-presenting cells (APCs). 11 IFN-β further induces regulatory T (Treg) cells and prevents the differentiation of T helper (Th)17 cells. 12 Direct effects on cluster of differentiation (CD) 80 and CD86 receptor expression on B cells are also observed, which leads to a modulation of their antigen-presenting function to T cells and inducing Th2 instead of Th1 cells. 13
Data in experimental neuritis: One study explored treatment with IFN-α/β protein before immunization in experimental autoimmune neuritis (EAN) and found attenuation of disease severity. 14 A second study used IFN-β at start of the immunization and with first clinical signs and described less inflammation in affected nerves. 15
Clinical data: In two randomized clinical trials (RCTs), IFN-β showed no beneficial effects in CIDP.16,17 One case report even indicated an exacerbation of CIDP after treatment of a MS patient with IFN-β. 18 There are also a few case reports of IFN-β-induced peripheral neuropathy in patients with MS that resolved with treatment cessation.19,20 The long-term safety data indicate, however, no neurotoxic potential. In MMN, IFN-β was used in non-randomized trials and showed moderate beneficial effects in three of nine patients in one study 21 and in four patients who failed other therapies. 22
Conclusion on AN relevance: Although not neurotoxic, there is no convincing evidence that IFN-β can be of potential benefit to CIDP or other AN.
Glatiramer acetate (GA)
GA is a random polymer of four amino acids that structurally resembles myelin basic protein (MBP) which is present in the central nervous system (CNS) and PNS. 23 GA competes with MBP in APCs for binding to the major histocompatibility complex (MHC) and thereby induces Th2 cells via secretion of interleukin-10 (IL-10).24–28 GA was also shown to elevate Treg and to decrease Th17 levels. 29 A more specific mode of action is the binding of GA and MHC to the T-cell receptor. It reacts as an antagonist of the immunogenic peptide MBP82–100. 30 MBP is essential for formation of CNS myelin. While it is also present in the PNS with identical amino acid sequences, it is non-essential for peripheral myelin formation since other proteins like P2 or P0 compensate lacking MBP.31,32 The above-mentioned mechanisms of GA mainly affect the peripheral immune compartments,33,34 even though there is some evidence that GA might be taken up by dendritic CNS cells. 35
Data in experimental neuritis: Two studies described beneficial effects of GA in EAN. Zhang et al. 36 injected GA daily subcutaneously (s.c.), before and during the induction phase of EAN. Aronovich et al. 37 administered GA intraperitoneally (i.p.), after onset of EAN. Clinical score improved but without significant effects on nerve conduction studies and histological assessment.
Clinical data: In CIDP patients with atypical manifestations, elevated MBP82–100 specific T-cell responses were found in low frequency. 38 Another study evaluated the activation of mononuclear cells specific for MBP in CIDP samples but found no specific activation pattern. 39 Case series or studies that evaluated the use of GA in patients with AN were not found.
Conclusion on AN relevance: Immunomodulatory effects of GA are mainly mediated through the resemblance to MBP. As MBP is non-essential for myelin formation in the PNS, GA is not an ideal candidate as a treatment option for patients with AN.
Dimethyl fumarate (DMF)
Fumaric acid esters were for a long time used in psoriasis and reports of coincidental amelioration of comorbid MS led to the conduction of first clinical trials in remitting-relapsing MS (RRMS). 40 To improve gastrointestinal tolerability, the drug was modified to DMF. 41 DMF is rapidly metabolized to the active metabolite monomethyl fumarate (MMF) that shifts the immune response to an anti-inflammatory Th2 subset with production of IL-4 and IL-5 in a dose-dependent manner.42,43 Increased overall Treg cell counts with a corresponding decrease in inflammatory Th17 cells were observed. 44 Activation of APCs is inhibited ex vivo and in vitro by reduced expression of costimulatory molecules and MHC II on B cells.43,45,46 Lymphopenia in DMF treatment preferentially affects CD8+ memory rather than CD4+ T cells.47–49 B memory cells are also reduced. 50 MMF upregulates expression of the nuclear factor erythroid 2-related factor 2 (Nrf2), which enables transcription of anti-oxidative genes in experimental autoimmune encephalomyelitis (EAE).51,52 MMF also shifts the balance of pro-inflammatory M1 and anti-inflammatory M2 macrophages to the latter. 53
Data in experimental neuritis: Three studies examined the effects of DMF in EAN. In a study by Pitarokoili et al., 54 rats were treated twice daily with 45 mg/kg body weight DMF orally. The authors demonstrated that DMF exerted immunomodulatory effects through the gut-associated lymphoid tissue by increasing Treg cell number and activation of Nrf2. Transfer of Peyer’s patches derived immune cells from DMF treated to treatment-naive rats during the induction phase of EAN significantly attenuated clinical, electrophysiological and histopathological signs of the neuritis. 54 Another study from the same group explored preventive effects of DMF in EAN. As a possible mode of action, induction of Nrf2 in axons was detected. 55 Han et al. 53 also detected anti-inflammatory effects of DMF in EAN in a preventive and a therapeutic paradigm. DMF treatment induced an anti-inflammatory shift from M1 to M2 macrophages. They also showed an increase in Nrf2 production in the nerves.
Clinical data: There are no RCTs or case reports with DMF or its variants in patients with CIDP or AN. There is no evidence of potential PNS neurotoxicity.
Conclusion on AN relevance: DMF mediates its immunomodulatory effects in the periphery, indicating a potential role for DMF as a treatment option. However, the overall immunosuppressant effects of DMF are not considerate strong. The common adverse effects such as flush, diarrhoea and nausea often cause a treatment switch in MS patients. 56 A promising and recently approved alternative is diroximel fumarate, a second-generation fumarate with bioequivalent active levels of MMF, which showed a reduced severity and frequency of gastrointestinal events. 57
Teriflunomide/leflunomide
Leflunomide is a prodrug which is metabolized to the active compound teriflunomide and its two isoforms. 58 Teriflunomide inhibits pyrimidine biosynthesis via the inhibition of the enzyme dihydroorotate dehydrogenase. The inhibition of the pyrimidine synthesis affects only rapidly proliferating cells, like antigen-stimulated lymphocytes.59,60 It induces a mild lymphopenia with absolute reduction of Th1 cells but not Th2 and Th17 cells. Therefore, a proportional increase in anti-inflammatory subsets can be expected.61,62 In EAE, teriflunomide showed a decreased infiltration of the CNS by macrophages, T cells and neutrophils.63,64 This is further supported by observed reduction of APCs in Peyer’s patches with an increase in Treg cells. Adoptive transfer of Treg cells from teriflunomide-treated mice, isolated from gut-associated lymphoid tissue, attenuated EAE severity. 65
Data in experimental neuritis: In one study, leflunomide was administered orally in dosages of 1.5, 10, 12.5 and 20 mg/kg daily during the induction phase of EAN, which are fourfold to 50-fold higher as used for rheumatoid arthritis treatment. 66 EAN was attenuated in all treatment groups. In a therapeutic setting, leflunomide 20 mg/kg daily p.o. halted disease progression. Histopathological studies showed a corresponding decrease in demyelination and cellular infiltration. 66 Similar results were seen in adoptive transfer EAN. 66
Clinical data: Teriflunomide was not explored in patients with immune-mediated neuropathy. However, a low incidence of mild-to-moderate peripheral neuropathy has been reported in patients with rheumatoid arthritis or MS, treated with teriflunomide.67–69 An in vitro screening on mouse embryonic stem cell–derived neurons found that high concentrations of teriflunomide lead to neurotoxicity, while neurodevelopment may be affected in undifferentiated cell lines in lower concentrations, which might be related to the teratogenic effects of teriflunomide in observed in vivo experiments.70,71
Conclusion on AN relevance: Teriflunomide and leflunomide as a prodrug are not appropriate due to the reported neurotoxicity.
Oral DMTs approved for highly active MS
Cladribine
Cladribine is a chlorated adenosine analogion. After cell uptake, it gets phosphorylated and is integrated in the cellular DNA and RNA. 72 Cladribine preferentially targets lymphocytes, which are more susceptible for integration of cladribine in their DNA and RNA due to higher levels of deoxycytidine kinase. It catalyses the rate-limiting step in the nucleotide salvage pathway, and opposing cytosol enzymes 5′-nucleotidases.73–75
Immunomodulatory effects: Cladribine induces a long-lasting depletion of autoreactive lymphocytes (approximately 45–33% compared with baseline after the end of the second cycle). 76 The effects are observed throughout all subpopulations. 73 The suppression of B cells is more pronounced compared with T-cell suppression. 76 Single-strand breaks lead to cell apoptosis via activation of p53 and through mitochondrial release of cytochrome c and apoptosis-inducing factor.77,78 The DNA incorporation of chlorated adenosine impairs cell repair mechanisms, protein transcription and gene expression. 77 Cladribine enhanced production of anti-inflammatory cytokines IL-4, IL-5 and IL-10.79,80
Data in experimental neuritis: We could not identify any studies investigating the use of cladribine in models of AN.
Clinical data: One case report describes the occurrence of GBS with positive anti-GD1b antibodies after exposure to cladribine. 81 Cladribine is therapeutically used in Waldenström macroglobulinemia that often goes along with a demyelinating peripheral neuropathy. 82 Fludarabine, another purine-analogion, showed some beneficial effects in patients with MAG neuropathy in a small study.83,84
A common concern of using nucleoside analogues is peripheral neuropathy. Regarding cladribine, in a study of 44 children with Langerhans cell histiocytosis treated with cladribine, none of the patient developed neuropathy. 85 With higher dosages, neurotoxicity was observed in 6 of 36 patients with refractory acute myeloid leukaemia.86,87 In the large phase III trials in MS, peripheral neuropathy was not increasingly observed. 88
Conclusion on AN relevance: The mainly observed immunomodulatory effects of cladribine are targeting peripheral lymphocytes and are exploited in the treatment of MS, indicative to be a good candidate for treating AN patients. This is encouraged by the data deriving from anti-MAG neuropathy patients. Neurotoxic effects were no concern in MS trials.
Functional sphingosine 1-phosphate receptor (S1PR) antagonists
Functional S1PR-antagonists exert immunosuppressive effects on CD4+ T cells.89,90 Fingolimod was the first one to be evaluated in MS.91–94 It reduces peripheral lymphocytes by impaired lymphocyte trafficking. 95 T cells seem to be the main cells affected by fingolimod, with intermediate effects on B cells and cells of the innate immune system.96–98 Fingolimod binds to different S1P-receptors. Effects on lymphocytes have been attributed to the S1PR1, while S1PR3 has been linked to the occurrence of atrioventricular block.99,100 Recently, more selective S1P-inhibitors that target S1PR1 and S1PR5 have been developed. Of those is siponimod the first oral medication approved for active secondary-progressive MS (SPMS), with a reduced risk of bradycardia. 101 Ozanimod and ponesimod have a similar mode of action as siponimod. Ozanimod was approved for RRMS, and ponesimod for active forms of MS, including active forms of SPMS.102,103 Other S1PR modulators currently invested are amiselimod and ceralifimod with a more selective modulation of the S1PR1. 99
Data in experimental neuritis: Zhang et al. 104 showed that fingolimod treatment nearly suppressed development of EAN, if administrated from the day of immunization, and reduced severity, when given with onset of symptoms. Fingolimod treatment led to a reduction in B and T cells as well as reduced macrophage infiltration of the sciatic nerve. Decreased Th17 cell proportion in peripheral blood, but an increased fraction in the lymph node, was also observed in EAN rats treated with fingolimod. 105 Ambrosius et al. reported EAN attenuation with oral fingolimod treatment (0.1 mg/kg body weight) in a preventive setting. Beneficial effects were also observed with fingolimod 1 mg/kg body weight i.p. in spontaneous autoimmune (poly-)neuritis (SAP) mice106,107 and oral treatment of 0.3 to 1 mg/kg body weight 108 in terms of reduction in disease severity, relapse rate and inhibition of demyelination, inflammation and axonal degeneration, while another study did not observe any effects in non-obese diabetic (NOD)-mice treated with 1 mg/kg daily i.p. 109 Schwann cells (SCs) promoting axonal regeneration under fingolimod stimulation were described for low concentrations, 110 while higher concentrations of fingolimod applied in cell culture models of dorsal root ganglia and SCs induce SC apoptosis and impede remyelination. 111
Clinical data: The FORCIDP trial investigated fingolimod in CIDP. Patients were randomized to either fingolimod 0.5 mg once daily or placebo in addition to standard treatment. 112 Overall, 106 participants were randomly assigned. The trial was ended after an interim analysis with worsening events in both groups, showing no superiority of fingolimod compared with placebo. Notably, S1PRs are expressed on the surface of peripheral and central neurons, as well as SCs; 111 however, there is no clinical evidence for potential neurotoxicity of S1PR antagonists.
Conclusion on AN relevance: Due to the negative results from the FORCIDP trial, S1PR antagonists will likely be of no relevance for AN treatment.
DMTs approved for highly active MS
Anti-CD20 antibodies
Up to date, three different anti-CD20 antibodies are established in the treatment of MS: rituximab, ocrelizumab and ofatumumab. Ofatumumab is the most recently approved anti-CD20 monoclonal antibody for RRMS and the only one given s.c., while the other two are given intravenously (i.v.). 113 In addition, while rituximab is a chimeric antibody and ocrelizumab a humanized anti-CD20 murine antibody, ofatumumab is the first immunoglobulin G (IgG)1 monoclonal anti-CD20 antibody that is fully human. 113 Ublituximab as another novel chimeric anti-CD20 antibody was recently evaluated in a phase II trial of 45 patients with RRMS showing promising results with 74% of the patients achieving no evidence of disease activity. 114 The epitopes differ between ofatumumab, rituximab, ublituximab and ocrelizumab. All four antibodies cause antibody-dependent cell lysis and complement-dependent cytotoxicity to CD20 expressing B cells. 115 The B-cell lineage expresses CD20 throughout all states of maturity and differentiation with the exception of plasma blasts, plasma cells and stem cells. Ocrelizumab, ofatumumab, rituximab and ublituximab induce cross-linking of CD20, which leads to an efficient activation of antibody-dependent cellular and complement-dependent cytotoxicity via the fragment crystallisable (Fc) domain.116,117 Ofatumumab strongly induces complement, while ocrelizumab has more antibody-dependent cellular cytotoxicity (ADCC) activity than rituximab. 115 The highest ADCC is shown by ublituximab. Its Fc domain has reduced fucose content, which results in an increased binding to the FcγIIIa receptor. 118 In addition, inebilizumab, a CD19-antibody targeting also early plasma cells, received approval for NMOSD.119,120
Data in experimental neuritis: There are several studies with conflicting results of B-cell depletion in models of autoimmune neuritis. Most of these are based on knockdown experiments or depletion of B cells via CD19 antibodies, which also target early plasma cells. Knock-down of CD4+ and CD8+ T cells ameliorated EAN, while B-cell knockout did not influence EAN course induced by P0180–199 peptide. 121 In SAP mice, P0-specific B cells and plasma cells were found to be increased and disease severity was attenuated by treatment with anti-CD19 antibodies. 122 However, another study reported that B-cell deficiency did not prevent SAP development. 123
Clinical data: Case reports and smaller series reported the beneficial use of anti-CD20 agents in therapy-refractory CIDP patients. 124 Some patients with CIDP harbour antibodies against nodal and paranodal proteins, and these patients tend to respond to rituximab but not to intravenous immunoglobulin (IVIg). 125 There is no evidence for PNS neurotoxicity of anti-CD20 or anti-CD19 agents. A phase II study showed promising results in untreated and relapsed Waldenström macroglobulinemia patients treated with ofatumumab monotherapy. 126 Two small, controlled studies investigated effects of rituximab in anti-MAG neuropathy patients; however, both studies failed to meet the primary endpoints using sensory scales.127,128 Secondary endpoints in the second study based on disability and time-to-walk scales, however, were met, and 40% improved under treatment. 128 Three case series or case reports also report the successful use of rituximab-monotherapy in non-IVIg-responsive MMN.129–131 As shown in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, rituximab might be a promising alternative for severe non-systematic vasculitic neuropathy (NSVN). 132
Conclusion on AN relevance: There is strong evidence for high relevance of anti-CD19/20 agents in the therapeutic armamentarium for AN. They are already used as off-label therapy in non-responders.
Natalizumab
Natalizumab is a humanized monoclonal antibody directed against an α4 integrin. It prevents interaction between very late antigen 4 (VLA4) and its ligand vascular cell adhesion molecule 1 (VCAM1) and inhibits thereby the migration of lymphocytes into the CNS.133,134 It is one of the most potent DMTs for MS. A well-characterized severe adverse effect is the – compared with other disease-modifying drugs – high incidence of progressive multifocal leukoencephalopathy (PML) under patients treated with natalizumab. 135 Therefore, before treatment initiation, anti–John Cunningham virus (JCV) seropositivity should always be checked and anti-JCV-positive patients should not be treated longer than 2 years or the JCV-copy status should be monitored. 136 Another emerging treatment strategy is to extend the dosage intervals of natalizumab to approximately 6 weeks. A randomized, open-label phase III trial confirmed effectiveness of natalizumab in patients when switched from standard dosage interval of 4 weeks to extended dosage intervals after more than 1 year of natalizumab treatment. 137
Data in experimental neuritis: Two studies examined the effects of antibodies against VLA4 in EAN. Preventive 138 and therapeutic 139 administration of natalizumab attenuated EAN course and histopathological inflammation.
Clinical data: One CIDP patient with treatment-resistant CIDP worsened after natalizumab treatment, even though T cells expressing α4 integrin were found in the inflamed nerve biopsy. 140 In a case series, three other treatment-resistant CIDP patients were successfully treated with an improvement in two and a stabilization in one patient. 141 There is no evidence of any neurotoxic potential of natalizumab.
Conclusion on AN relevance: Further studies would be needed to investigate the role of natalizumab as a treatment option. The high incidence of PML, which increases significantly after 2 years, limits its potential. 142
Currently investigated DMTs
Tyrosine kinase inhibitors (TKIs)
TKIs were initially designed for tumour therapy, for example, imatinib, which inhibits ABL-tyrosine kinase in chronic myeloid leukaemia. Many different TKIs are currently under clinical evaluation or already integral part of tumour treatment. Bruton’s tyrosine kinase (BTK) is involved in B-cell receptor signalling. BTK inhibitors (BTKIs) prevent B-cell maturation without wiping out resident B cells. BTK is essential for B-cell maturation, and the lack of the enzyme is responsible for the development of x-linked agammaglobulinemia.143,144 It controls the progression of pre-B cells and the entry of B cells into follicular structures.145,146 Furthermore, activation of mature B cells and their termination into memory or plasma cells is impaired. 147 Two brain-penetrant BTKIs have been tested in MS. Tolebrutinib showed in a phase II trial promising results on safety and on radiological disease activity. 148 Evobrutinib was also tested in a phase II trial for dosage and safety profile against placebo and DMF. 149 Tolebrutininb and evobrutinib are irreversible inhibitors of BTK. 150 In EAE, a dose-dependent effect of evobrutinib was seen, with reduced expression of antigen-presentation molecules on B cells and a reduced development of encephalitogenic T cells. 151 This led the pathway for the evaluation of other BTKIs in MS. Fenebrutinib, currently evaluated in a phase II trial (NCT05119569), 152 and remibrutinib are reversible BTKIs,153,154 while orelabrutinib is another irreversible BTKI evaluated in a phase II trial (NCT04711148).153,155 Overall, there are nine phase III trials ongoing, six for RRMS against teriflunomide and three in primary-progressive MS (PPMS)156,157 and SPMS. 158 The results should further enlighten the role of BTKs for all forms of MS. Zanubrutinib is another irreversible BTKI tested in a phase II trial in NMOSD (NCT05356858). 159 In addition, tolebrutinib is tested in patients with generalized myasthenia gravis. 160
Data in experimental neuritis: To our knowledge, no studies investigated the role of TKIs or BTKIs in models of AN.
Clinical data: There are no clinical data of the use of BTKIs or other TKIs in CIDP. However, TKIs are used for treatment of patients with chronic lymphatic leukaemia and Waldenström macroglobulinemia. A case series reported the successful treatment of patients with anti-MAG neuropathy with ibrutinib. 161 Some TKIs, for example, vascular endothelial growth factor receptor TKI, may induce sensory neuropathy;162,163 however, neurotoxicity was not reported in phase II MS trials with TKIs.148,149 A phase II trial is currently evaluating the safety of a combination therapy of an anti-CD20 agent with acalabrutinib, a BTKI, in patients with Waldenström macroglobulinemia, IgM monoclonal gammopathy of undetermined significance (MGUS) and anti-MAG neuropathy (NCT05065554). 164
Conclusion on AN relevance: There is some evidence that BTKIs may be useful in AN. The potential reversible effects of BTKI inhibition after treatment discontinuation are a major advantage during B-cell depletion. The existence of long-term data of different BTKIs with many patients from leukaemia treatment could accelerate the clinical translation. Hence, no penetration of the blood–brain barrier (BBB) is needed; more BTKIs could be applicable for AN treatment.
Laquinimod
Laquinimod is an orally available quinoline-3-carboxamide. 165 Laquinimod slowed progression of disability and reduced rate of relapse in RRMS. 166 However, it received a negative review by the European Medicines Agency due to concerns regarding long-term exposure with higher occurrence of cancers and its teratogenic potential in animal studies. 167 The exact mode of action of laquinimod is currently unknown. Several studies point to an activation of the aryl hydrocarbon-receptor pathway and a dependency of laquinimod on this receptor to mediate anti-inflammatory effects.168–170 These effects are a decreased Th1 and Th17 response with an increased reactivity of Treg cells. Neuroprotective effects are linked to other mechanisms, for example, upregulation of brain-derived neurotrophic factor receptors. 171
Data in experimental models of neuritis: Two studies showed that laquinimod attenuated EAN. Zou et al. 172 compared the effects of laquinimod in different concentrations (0.16, 1.6 and 16 mg/kg s.c.) to its predecessor linomide (16 mg/kg s.c.) and sham treatment. Dose-related reduction in EAN severity was shown with correlating reduced cellular infiltration and a shift towards anti-inflammatory Th2 cell response. Pitarokoili et al. 173 showed similar positive results with daily treatment of 12.5 or 25 mg/kg orally.
Clinical data: To our knowledge, no clinical studies examining the effects of laquinimod on AN were performed or are currently conducted. There is also no evidence for PNS neurotoxicity.
Conclusion on AN relevance: Due to the limited data and the unknown exact mode of action, the role of laquinimod for AN treatment is unclear.
DMTs for NMOSD
IL-6-receptor antagonists
Satralizumab and tocilizumab are monoclonal antibodies against IL-6 receptors. Off-label uses of tocilizumab in NMOSD as mono- or add-on medication were shown to be effective to reduce disease activity and progression in myelin oligodendrocyte glycoprotein (MOG-) or aquaporin-4- (AQP4-) positive and negative patients.174–177 Satralizumab was engineered with an enhanced plasma resistance, 178 and it is given s.c. in contrast to tocilizumab, which is administered i.v. 177 Satralizumab was tested in two phase III trials as add-on treatment or as monotherapy and reduced relapses in both trials. Primary endpoints were the time to and incidence of a relapse in both trials and satralizumab led to a risk reduction of 55% 178 and 65%, respectively. 179 No significant difference in the secondary endpoints (change in Visual Analogue Scale pain score and the Functional Assessment of Chronic Illness Therapy fatigue score from baseline to week 24) was noted. It was generally well tolerated.178,179 The antibodies inhibit the binding of IL-6 to its receptor. There is clinical and experimental evidence that IL-6 plays a key role in the pathogenesis of NMOSD.180–183
Data in experimental neuritis: EAN is associated with increased IL-6 levels during disease onset. 184 Other studies hint to neuroprotective effects of IL-6 as its nasal application ameliorated disease severity in EAN. 185
Clinical data: IL-6 may be increased in the cerebrospinal fluid (CSF) of CIDP patients, while serum IL-6 levels are normal. 186 Another study could find no significant differences in IL-6 CSF expression. 187 Nerve biopsies of the sural nerve showed an upregulation of IL-6 in CIDP patients associated with upregulated neurotrophic growth factor receptors, indicating a role in nerve regeneration. 188 There is no evidence of any neurotoxic potential of satralizumab and tocilizumab.
Conclusion on AN relevance: The role of IL-6 in AN pathogenesis appears to be minor, and therefore, IL-6-receptor antagonists are not relevant for repurposing in AN patients.
Eculizumab and other anti-complement agents
Eculizumab is a humanized monoclonal antibody that is approved for NMOSD in AQP4-positive patients. 120 Ravulizumab is another terminal complement protein (C5) inhibitor, which is currently evaluated in NMOSD. 189 Due to drug modifications, it has – compared with eculizumab – a prolonged half-life, which extends the dosage intervals. 190 Eculizumab binds to C5 and prevents cleavage to C5a and C5b and thereby inhibiting membrane attack complex (MAC) formation. MAC is the final step of the complex cascade and is able to mediated cell lysis. 191 Both soluble C5a and MAC can be detected in plasma and CSF of NMOSD patients. 192
Data in experimental neuritis: In EAN, complement depletion mitigates clinical course and nerve injury.193–195 The passive transfer of the disease with pathogenic IgG with complement C3 reactivity from CIDP patients into rats, results in nerve conduction impairment and demyelination 196 and suppression of disease following administration of soluble complement receptor one (sCR1), indicates potential association of complement in CIDP. Furthermore, deposition of complement components on nerves 197 and of complement-fixing autoantibodies on the myelin sheath and high systemic complement levels that correlated with disease severity 198 further point to an essential role of complement in the pathogenesis of CIDP and GBS.199,200
Clinical data: There is no evidence of any neurotoxic potential of eculizumab. We found a phase II open-label study that is currently investigating potential effects on CIDP patients treated with a C1s inhibitor, which is part of the activation of the classical complement pathway (NCT04658472). 201 Eculizumab was used as an add-on therapy to IVIg in MMN patients to prove short-term safety in an open-label study. Reduction of IVIg dosage was not significant, but secondary outcome measures showed promising results like a small, but significant decrease in conduction block. 202 A randomized phase II study recently started to further examine the role of complement in MMN by the use of a C2 inhibitor, which plays a role in both the classical and lectin pathway of complement activation (NCT05225675). 203 Eculizumab was further evaluated in two phase II studies of GBS patients as add-on therapy to IVIg and showed inconclusive results. In one study, a benefit was observed in secondary outcomes, 204 while the other study could randomize only eight patients with severe GBS. 205 However, both showed the safety of eculizumab infusions. A phase III trial is currently ongoing (NCT04752566). 206 ANX005 inhibits C1q and is another classical complement inhibitor currently evaluated in GBS in a phase II/III trial (NCT04701164). 207
Conclusion on AN relevance: There is strong evidence that anti-complement agents are highly relevant in CIDP treatment as indicated by the role of complement in CIDP pathogenesis and are also relevant for MMN and GBS.
DMTs for other autoimmune disorders
Even though out of the scope of this review, we would like to mention another drug class with highly promising potential to impact AN disease course, which are inhibitors of the neonatal Fc-receptor (FcRn). FcRn inhibitors are currently evaluated in myasthenia gravis and excellently reviewed elsewhere. 208 In short, the neonatal Fc-receptor protects IgGs from lysosomal degradation and enables re-entering of IgGs into the circulation. In autoimmune disease with pathogenic antibodies, an inhibition of the receptors leads to a faster degradation of those autoantibodies. Several FcRn agents were evaluated in myasthenia gravis and showed promising results. Recently, efgartigimod (i.v.), a humanized IgG1 Fc fragment, was approved for acetylcholine receptor antibody-positive myasthenia gravis after a phase III trial with significant clinical improvement in the patients as measured by Myasthenia Gravis Activities of Daily Living (MG-ADL) after 8 weeks. Rozanolixizumab is an s.c. administered anti-FcRn IgG4 monoclonal antibody currently in a phase III trial in generalized myasthenia gravis (NCT04650854). 209 Nipocalimab, given i.v., is another IgG1 anti-FcRn monoclonal antibody currently evaluated in a phase III trial (NCT04951622). 210 All three inhibitors led to a meaningful reduction of 70–90% of circulating IgG levels. 208 A fourth FcRn inhibitor, batoclimab, is recently recruiting patients for a phase III trial (NCT05403541). 211 In MOG-associated EAE, treatment with an FcRn-antibody ameliorated disease severity. 212
Data in experimental neuritis: We could not identify any specific studies evaluating FcRn inhibitors in autoimmune neuritis models.
Clinical data: There was no increased neurotoxic potential reported for the FcRn inhibitors. Currently one FcRn inhibitor – efgartigimod – is evaluated in a phase II trial in CIDP (NCT04281472). 213 Another phase II/III trial is planned for the evaluation of nipocalimab, a third FcRN inhibitor (NCT05327114). 214
Conclusion on AN relevance: FcRn is a promising therapeutic target in both CIDP and anti-MAG neuropathy. The reduction is not equally distributed throughout all subclass, with a less prominent effect on IgG4, which is of great relevance in IgG4-related nodopathies. 208 Inhibition of FcRn is especially of interest for AN patients, as the IgG reduction is only transient without altering the function of plasma cells and memory B cells and, therefore, not increasing the risk of infections. 215
Discussion
The term ‘drug repurposing’ is not clearly defined 216 and includes a variety of different strategies ranging from hypothesis-driven approaches, that is, finding a new indication for a licensed drug, up to computational approaches by use of ‘signature mapping’, or pathway ‘matching’ 217 to identify new indications out of the original scope of the drug. Repurposing licensed drugs is an attractive option particularly for rare diseases like CIDP where the pathogenesis is incompletely understood and studies are difficult to conduct, resulting in a high risk of failure. As a general limitation, it should be noted that DMTs for RRMS or NMOSD have been evaluated in a mainly well-defined and homogeneous clinical condition with well-established clinical outcome parameters (i.e. annual relapse rate) and specific laboratory parameters (i.e. non-invasive imaging biomarkers, like magnetic resonance imaging). This is, however, not the case for SPMS and PPMS with less well-established primary and secondary endpoints. Using the same drug in AN means applying a drug to a novel condition with a more diverse clinical presentation, different immunopathogenesis and heterogeneous or less well-defined quantitative outcome measures. Furthermore, regulatory concerns such as pre-existing patents that could impede commercialization of the repositioned drug need to be considered.
Notably, most of the presently discussed DMTs are established in RRMS or SPMS, as only ocrelizumab is currently approved as a DMT for PPMS and the other DTMs showed only limited treatment success. 218 Many CIDP patients also present with a pattern resembling a ‘primary-progressive’ disease course, with clinical features like PPMS. The heterogeneity of AN patient cohorts is also a consideration in the context of the translational therapeutic failures noted with some DMTs that may reflect different disease subtypes.219,220 This is highlighted by the fact that there are several clinical variants of CIDP including pure motor or sensory forms, multifocal motor-sensory neuropathy (Lewis–Sumner syndrome) and pure distal forms. Some patients only respond to steroids, while others show a better response to IVIg suggesting differences in the immunopathogenesis of the disease. 219
Several pathogenic factors which are the target for DMTs in NMOSD and MS are shared in AN, especially CIDP: production of immunogenic antibodies against a known or unknown antigen by B cells, infiltration of inflammatory cells like macrophages, disruption of the BBB or BNB, respectively, as wells as complement activation, a pathological hallmark in NMOSD (Table 1 and Figure 1). In both, AN and MS/NMOSD, inflammatory attacks induce demyelination and axonal damage. Chronical demyelination and axonal degeneration are believed to be the main driver of disease progression in PPMS and SPMS, but also in some CIDP patients. 221
The question as to which of the discussed drugs promise the highest benefit is, therefore, challenging especially since there is no standardization for this evaluation process or adequately weighing experimental and clinical data. Moreover, factors such as potential neurotoxicity, time to improvement and the possibility of the desired drug to be used as add-on therapy may further influence the selection process. Accordingly, by applying these criteria, many of the DMTs can be already excluded. Interferons and fingolimod are not applicable because of the negative evidence-based data of RCTs.16,17,112 GA also is not suitable, as its mechanism of action is based on its resemblance to MBP, which is a non-relevant antigen in EAN and AN. 39 Teriflunomide may exert a beneficial mode of action, but its weak immunopathogenic effect relevant to CIDP and other AN and the possibility of association with peripheral neuropathy are not favourable factors. Likewise, although there are promising preclinical data that suggest laquinimod may work for CIDP, the unresolved issue of long-term safety will prevent its further consideration as a potential treatment option for AN. Experimental data also suggest a beneficial effect of natalizumab; however, the available clinical data and its safety profile with the risk of development PML in JC-virus-positive patients 142 probably argue against its further exploration in AN.
In contrast, cladribine seems to be a good candidate for a trial in AN patients. This view is also supported by promising results in anti-MAG neuropathy patients. Anti-CD20 agents also appear attractive, based on the pivotal role of certain autoantibodies, such as MAG and gangliosides, and the clinical experience with rituximab as off-label drug. At present, a controlled study with rituximab has started in Italy for CIDP patients. 222 The anti-CD20 antibodies differ as mentioned by their targeted epitope and their potential to activate ADCC. As B-cell depletion is described sufficiently and reliable for all agents, and no head-to-head studies are available, similar translational results to CIDP should be expected. Anti-CD19 therapies like inebilizumab also target mature B cells like anti-CD20 agents; however, CD19 is expressed throughout B-cell maturation and also on secreting plasmablasts and plasma cells. 223 Hence, anti-CD19 agents might provide an additional benefit by directly targeting the sources of pathogenic autoantibody production, plasmablasts and plasma cells. Obexelimab is another antibody on the horizon, targeting CD19 along with the inhibitory FcγRIIb receptor. 224 Obexelimab is designed to inhibit B-cell function without destroying the immune cells, 225 potentially reducing susceptibility to infections as seen by long-lasting B-cell depletion. Comparative clinical studies regarding autoimmunity of the CNS are currently missing to support this hypothesis. The strong biological plausibility and the comparable side-effect profile compared with the anti-CD20 agents should strongly encourage RCTs in AN, especially in the IgG4-related nodopathies which do not respond to IVIg. 226 A compelling alternative to anti-CD20 antibodies, despite the lack of preclinical data, are BTKIs. The potential reversibility after treatment discontinuation and the well-tolerability as well as the low incidence of opportunistic infections encourages further exploration for AN.
Preclinical and clinical data regarding complement also imply a potential role of anti-complement agents. Advantageously, data from an RCT already demonstrated that the parallel treatment of IVIg and eculizumab does not diminish efficacy of anti-complement activity. 204 Complement is, however, no treatment target in IgG4-nodopathy CIDP patients as they do not cause complement-fixing or macrophage-mediated demyelination.226,227 The role of IL-6 and hence the use of IL-6 receptor antagonists are not well explored, and there is no evidence in favour or against its use in CIDP or other AN.
Taken together, repurposing DMTs, which are well established in MS and NMOSD, is an attractive option for AN to expand the therapeutic landscape, provided a careful selection process has been employed implementing objective assessment of the available clinical and experimental data. Based on careful comparative clinical and safety assessments, encouraging data for new clinical trials in AN derive from B cell–targeted therapies (either antibodies or novel approaches such as BTK inhibition), complement-targeting DMTs and cladribine. The anti-FcRn inhibitors, currently explored in both NMOSD and CIDP, may be additional options.