
Abstract
In the past few years, acquired demyelinating syndromes of the central nervous system associated with antibodies against myelin oligodendrocyte glycoprotein (MOG) have evolved into a new inflammatory disease entity distinct from neuromyelitis optica spectrum disorders or multiple sclerosis. The meticulous clinical description of patients with MOG IgG antibodies (MOG-IgG) has been achieved by development and use of highly specific cell-based assays. MOG-IgG associated disorders comprise a wide spectrum of syndromes ranging from acute disseminated encephalomyelitis predominantly in children to optic neuritis or myelitis mostly in adults. In recent studies, phenotype of MOG-IgG associated disorders has further broadened with the description of cases of brainstem encephalitis, encephalitis with seizures and overlap syndromes with other types of autoimmune encephalitis. In this review, we provide an overview of current knowledge of MOG-IgG associated disorders, describe the clinical presentations identified, highlight differences from neuromyelitis optica spectrum disorders and multiple sclerosis, summarize clinical outcome and concepts of immune treatment, depict the underlying mechanisms of antibody pathogenicity and provide the methodological essentials of MOG-IgG assays.
Keywords
Introduction
Myelin oligodendrocyte glycoprotein (MOG) is a highly conserved protein that is exclusively expressed in oligodendrocytes in the central nervous system (CNS).1,2 Multiple isoforms of MOG exist that have identical extracellular immunoglobulin (Ig) domains, but differentially spliced intracellular C-termini. The differences in the C-terminal amino acids are the basis to distinguish α or β isoforms of MOG.3,4 Knowledge about the biological role of MOG and its isoforms is limited; however, the encephalitogenic potential of MOG that is eliciting demyelinating immune responses has been demonstrated in numerous experimental models.5–7 For this reason, MOG-IgG antibodies (MOG-IgG) were extensively studied in the last two decades in different acquired demyelinating syndromes (ADSs). The development and use of highly specific cell-based assays (CBAs) enabled the description of a variety of clinical disease manifestations ranging from certain ADS, for example, acute disseminated encephalomyelitis (ADEM) predominantly in children or optic neuritis mostly in adults, to cases of encephalitis with seizures.8–11 This broad spectrum of clinical phenotypes associated with MOG-IgG has evolved into a new inflammatory CNS disease entity that is distinct from both multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSDs).
In this review, we provide an overview of current knowledge of MOG-IgG associated disorders, describe the clinical presentations identified, highlight differences from NMOSD and MS, summarize clinical outcome and concepts of immune treatment, depict the underlying mechanisms of antibody pathogenicity and provide the methodological essentials in MOG-IgG assays.
Clinical aspects
Frequency of MOG-IgG in ADSs
The majority of studies of MOG-IgG in ADS have been retrospective and included selected patient populations. Only a few of the latest studies were designed prospectively or used a population-based approach. MOG-IgG were detected in 65 (31%) of 210 children with ADS in a large study conducted in Austria and Germany, 12 in 76 (32%) of 237 children in a UK study, 13 in 31 (22%) of 151 children in a study from The Netherlands, 14 in 94 (39%) of 239 children in a study from Spain, 15 in 17 (18%) of 92 children in a study from Denmark 16 and in 84 (31%) of 274 children in a study from Canada. 17 Reported incidence rates ranged from 0.16 to 1.4 per 100,000 that is – among the group of ADS – in the range of NMOSD, but clearly below the incidence of MS (see the following).12,18,19
Summarizing the data from all available studies that used CBAs [combined with fluorescence activated cell sorting (FACS) or immunofluorescence (IF)] in aquaporin-4 (AQP4)-IgG negative non-MS ADS to analyse MOG-IgG revealed a clear association of their prevalence with age. The proportion of patients with MOG-IgG positive ADS was higher among children (39%)13–15,17,20–35 than among mixed cohorts of children and adults (29%)36–51 or adults (23%)8,12,52–75 [Figure 1(A)]. This higher seroprevalence of MOG-IgG in children might be a consequence of the age-dependent manifestation of different demyelinating CNS diseases, as MS and AQP4-IgG positive NMOSDs are more common in adults.76,77

Frequency and clinical presentation of MOG-IgG associated disorders are age-dependent. (A) Frequency of MOG-IgG in AQP4-IgG negative non-MS demyelinating diseases out of studies including children,13–15,17,20–35 mixed cohorts of children and adults36–51 and adults8,12,52–75 is shown. (B) Frequency of the main clinical phenotypes (ADEM, optic neuritis, myelitis) out of studies including children13–15,17 and adults10,18,78 is shown.
Spectrum of MOG-IgG associated demyelinating syndromes
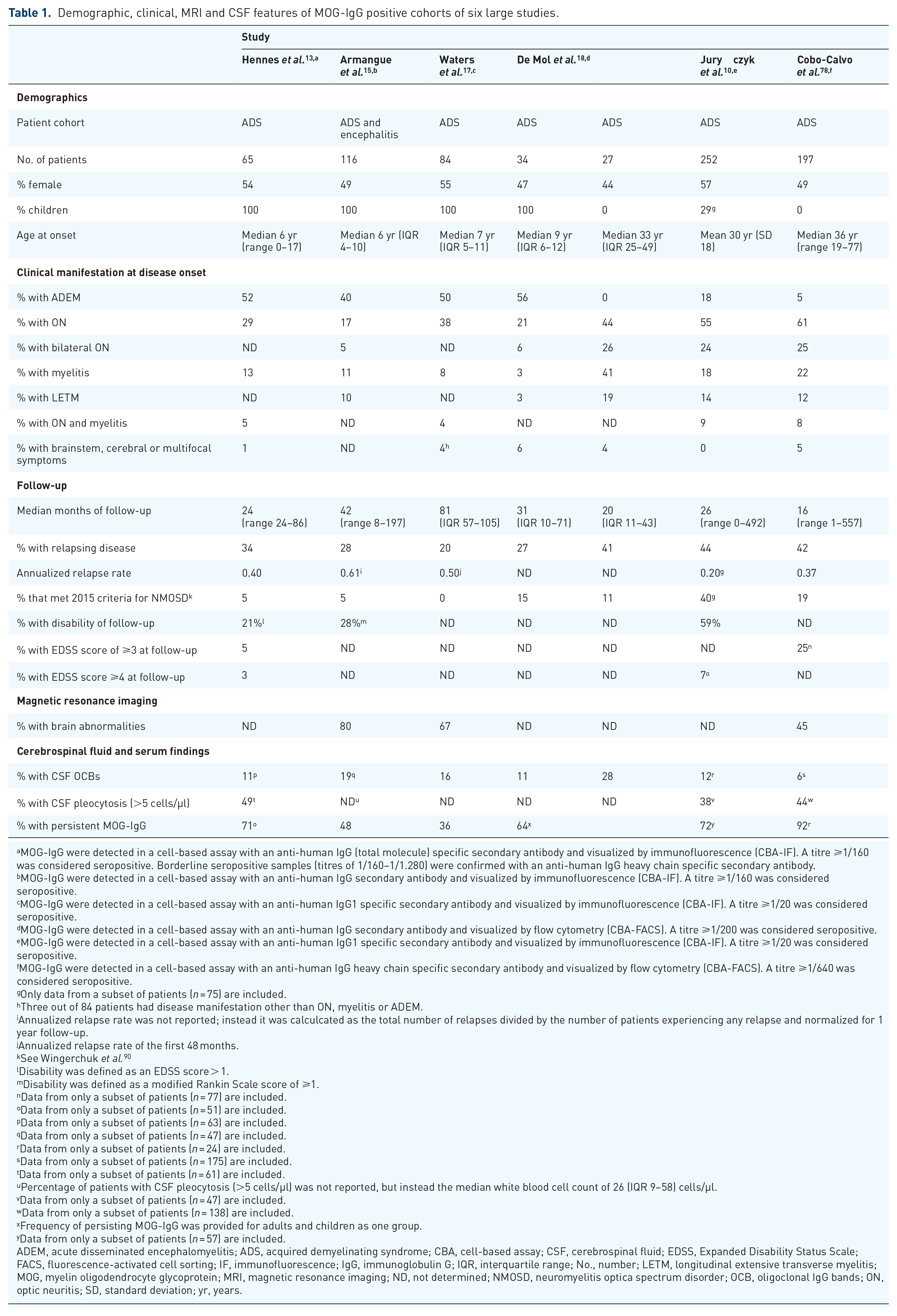
Clinical and magnetic resonance imaging (MRI) characteristics of patients with positive MOG-IgG were reported by a multitude of studies in the past few years including different cohorts of patients, that is, children, adults or both, patients with monophasic and recurrent or with relapsing disease course only.9,10,13,14,17,23,25,35,48,49,51,56,57,68,78–86 Overall, the clinical phenotype of MOG-IgG associated demyelinating syndromes changes with age from ADEM-like (ADEM, ADEM–optic neuritis, multiphasic disseminated encephalomyelitis) in children to opticospinal (optic neuritis, myelitis) in adults. A detailed overview on six recent representative large observational studies showing the frequency of different disease manifestations in adults and children is given in Table 1. Accordingly, MOG-IgG positive children presented as ADEM in approximately 50% of cases, whereas adults in less than 10%. Vice versa, onset with optic neuritis or myelitis was observed in up to 50% and 30% of adults, whereas in approximately 25% and 10% of children (Figure 1(B)]. Manifestations with brainstem, cerebral or multifocal symptoms were – in most studies – quite infrequent (<10%) at any age.10,13,15,17,18,78,87 Symptoms associated with involvement of the area postrema, for example, nausea, vomiting and hiccups, were reported in a small subset of patients (at or before presentation). The majority of these patients did not show discrete area postrema lesions, but patchy, poorly demarcated lower brainstem lesions (most commonly in the context of ADEM; the isolated presentation was rare).87–89
Demographic, clinical, MRI and CSF features of MOG-IgG positive cohorts of six large studies.
MOG-IgG were detected in a cell-based assay with an anti-human IgG (total molecule) specific secondary antibody and visualized by immunofluorescence (CBA-IF). A titre ⩾1/160 was considered seropositive. Borderline seropositive samples (titres of 1/160–1/1.280) were confirmed with an anti-human IgG heavy chain specific secondary antibody.
MOG-IgG were detected in a cell-based assay with an anti-human IgG secondary antibody and visualized by immunofluorescence (CBA-IF). A titre ⩾1/160 was considered seropositive.
MOG-IgG were detected in a cell-based assay with an anti-human IgG1 specific secondary antibody and visualized by immunofluorescence (CBA-IF). A titre ⩾1/20 was considered seropositive.
MOG-IgG were detected in a cell-based assay with an anti-human IgG secondary antibody and visualized by flow cytometry (CBA-FACS). A titre ⩾1/200 was considered seropositive.
MOG-IgG were detected in a cell-based assay with an anti-human IgG1 specific secondary antibody and visualized by immunofluorescence (CBA-IF). A titre ⩾1/20 was considered seropositive.
MOG-IgG were detected in a cell-based assay with an anti-human IgG heavy chain specific secondary antibody and visualized by flow cytometry (CBA-FACS). A titre ⩾1/640 was considered seropositive.
Only data from a subset of patients (n = 75) are included.
Three out of 84 patients had disease manifestation other than ON, myelitis or ADEM.
Annualized relapse rate was not reported; instead it was calculcated as the total number of relapses divided by the number of patients experiencing any relapse and normalized for 1 year follow-up.
Annualized relapse rate of the first 48 months.
See Wingerchuk et al. 90
Disability was defined as an EDSS score > 1.
Disability was defined as a modified Rankin Scale score of ⩾1.
Data from only a subset of patients (n = 77) are included.
Data from only a subset of patients (n = 51) are included.
Data from only a subset of patients (n = 63) are included.
Data from only a subset of patients (n = 47) are included.
Data from only a subset of patients (n = 24) are included.
Data from only a subset of patients (n = 175) are included.
Data from only a subset of patients (n = 61) are included.
Percentage of patients with CSF pleocytosis (>5 cells/µl) was not reported, but instead the median white blood cell count of 26 (IQR 9–58) cells/µl.
Data from only a subset of patients (n = 47) are included.
Data from only a subset of patients (n = 138) are included.
Frequency of persisting MOG-IgG was provided for adults and children as one group.
Data from only a subset of patients (n = 57) are included.
ADEM, acute disseminated encephalomyelitis; ADS, acquired demyelinating syndrome; CBA, cell-based assay; CSF, cerebrospinal fluid; EDSS, Expanded Disability Status Scale; FACS, fluorescence-activated cell sorting; IF, immunofluorescence; IgG, immunoglobulin G; IQR, interquartile range; No., number; LETM, longitudinal extensive transverse myelitis; MOG, myelin oligodendrocyte glycoprotein; MRI, magnetic resonance imaging; ND, not determined; NMOSD, neuromyelitis optica spectrum disorder; OCB, oligoclonal IgG bands; ON, optic neuritis; SD, standard deviation; yr, years.
Delineating MOG-IgG associated disorders from other demyelinating CNS diseases
Patients with MOG-IgG associated demyelinating syndromes show certain demographic and clinical characteristics, cerebrospinal fluid (CSF) and MRI findings that allow the differentiation from patients with MS or APQ4-IgG positive NMOSD to a certain extent and can be the basis for reasonable antibody testing. A comprehensive comparison between MOG-IgG associated disorders, APQ4-IgG positive NMOSD and MS is shown in Table 2; MRI findings are further detailed in the following.
Demographic, clinical, MRI and CSF features of MS, AQP4-IgG positive NMOSD and MOG-IgG associated disease.

Apart from the references provided in the table, the data summarize serveral studies of MS,76,101–103 AQP4-IgG positive NMOSD80,90,104–106 and MOG-IgG associated disorders.9,10,13,14,23,25,35,48,49,51,56,57,68,78,79,81–86,107
ADEM, acute disseminated encephalomyelitis; AQP4, aquaporin-4; CSF, cerebrospinal fluid; CSF, cerebrospinal fluid; IgG, immunoglobulin G; MDEM, multiphasic disseminated encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; MS, multiple sclerosis; MRI, magnetic resonance imaging; NA, not available; NMOSD, neuromyelitis optica spectrum disorder (according to the 2015 diagnostic criteria); OCB, oligoclonal IgG band; pRNFL, peripapillary retinal nerve fibre layer.
MRI
Brain MRI in patients with MOG-IgG associated disorders typically shows few, poorly demarcated, ‘fluffy’, sometimes large lesions that affect both the white matter and the grey matter, the latter including cortex and deep grey nuclei.9,91 However, a reliable discrimination of MOG-IgG associated disorders from AQP4-IgG positive NMOSD and MS based on brain MRI findings is not possible.9,10,13,15,17,18,78,91,95,108 Characteristic MRI features have been reported for optic pathway and spinal cord. Patients with MOG-IgG associated disorders and optic neuritis typically had bilateral involvement of the anterior optic pathways with long lesions and optic nerve head swelling.95,109 Bilateral involvement was of similar frequency in MOG-IgG associated disorders and AQP4-IgG positive NMOSD, but significanly more frequent than in MS. Affection of the chiasma was reported in some patients with MOG-IgG associated disorders; whether the frequency differs from AQP4-IgG positive NMOSD is still contradictory. The involvement of the optic tract was uncommon in MOG-IgG associated disorders.95,109 MRI of the spinal cord typically showed longitudinally extensive T2-signal abnormalities (>3 vertebral segments) predominantly of the thoracolumbar region 40 involving the ventral spinal cord parenchyma confined to the grey matter (sagittal line and axial H sign) without contrast-enhancement, 93 allowing distinction of myelitis from APQ4 positive NMOSD and MS. Longitudinally extensive T2 lesions were of similar frequency in MOG-IgG associated disorders and AQP4-IgG positive NMOSD but not found in MS.93,94 Multiple spinal cord lesions and conus involvement were more frequent with MOG-IgG than AQP4-IgG but not different from MS. 93 Besides these reported MRI features, the initial scan of the brain15,17,78,85 as well as of the spinal cord 110 can be normal in patients with MOG-IgG associated disorders, also despite severe clinical manifestation, which can lead to diagnostic uncertainty.
Expanding the MOG-IgG disease spectrum
Encephalitis
The association of MOG-IgG with an encephalitic presentation has been known since the first report in 2017. An adult was described with steroid-responsive encephalitis involving the cortical areas who experienced focal seizures that subsequently generalized and who was eventually shown to be positive for MOG-IgG. When the patient’s treatment of prednisolone was tapered, a relapse occurred with optic neuritis. 111 Later case series confirmed the association between MOG-IgG, cortical brain lesions on MRI (typically with contrast enhancement) and seizures. The vast majority of patients had a relapsing disease course and had experienced demyelinating events such as optic neuritis or myelitis before, with or after encephalitis onset; and some patients had additional deep white matter or brainstem lesions on MRI.78,112–115
To further explore the significance of encephalitis associated with MOG-IgG, a large, prospective, multicentre, observational study was performed that included children with the whole spectrum of encephalitis (i.e. of infectious, autoimmune and unknown origin). The authors reported that among patients with autoimmune encephalitis (other than ADEM) MOG-IgG were more common (34%) than all neuronal antibodies combined (with N-methyl-D-aspartate-receptor [NMDAR] antibodies in 22% as the second most frequent). 15 These MOG-IgG positive encephalitis patients developed clinical syndromes including decreased level of consciousness (100%), seizures [64% (45% with status epilepticus)], fever (59%), and abnormal behaviour (50%) and movements (36%). Brain MRI showed extensive cortical involvement, basal ganglia or thalamic involvement, in some cases also only minimal changes (those associated with refractory status epilepticus), or revealed normal scans. Within a median follow-up of 45 months, 23% of patients had at least one relapse; almost all of them showed a demyelinating syndrome (optic neuritis or myelitis). 15
Altogether, MOG-IgG were found to be associated with the clinical presentation of encephalitis (other than ADEM). The majority of patients showed also features of demyelination either clinically before, concomitantly or after encephalitis onset, or by means of MRI.
Overlap syndromes
Overlap syndromes with MOG-IgG and NMDAR antibodies are known. A recent study reported that in patients with NMDAR encephalitis concurrent glial antibodies were present in approximately 4% of cases; half of them were MOG-IgG. 116 NMDAR encephalitis patients with additional MOG-IgG showed more frequently atypical disease manifestation including brainstem or cerebellar symptoms or presented distinct demyelinating features such as optic neuritis or myelitis; MRI revealed lesions typically in subcortical white matter, infratentorial region or the spinal cord with facultative contrast-enhancement. In some cases involvement of the basal ganglia and cortical regions as well as meningeal enhancement have been reported.47,116 From a pathophysiological point of view, the shared contribution of NMDAR antibodies and immune responses to myelin dysfunction is unknown, but it should be noted that oligodendrocytes contain NMDAR. 117 It has been suggested that MOG-IgG could reflect, for example, a secondary immune reaction. 118 However, patients with overlapping antibodies also often had a history of episodes of encephalitis or demyelinating syndromes.47,116 Of note, the distribution of the concurrent antibodies was different in serum and CSF in some patients, suggesting different compartmentalization of the immune responses. 116 Evolution and sequence of concurrent antibodies as well as the underlying immune mechanisms have to be explored by further studies. Nevertheless, these findings emphasize the importance of examining MOG-IgG in patients with NMDAR encephalitis and atypical clinical or imaging features, especially because double-positive patients might have worse prognosis and possibly need more aggressive immune treatment. 47
Seizures
Seizures as an isolated phenomenon have been reported to be associated with MOG-IgG, that is, seizures were not accompandied by clinical manfestation of encephalitis or ADS, and brain MRI was unremarkable at onset. Seizures occurred solitarily or as cluster over a short interval of days. As a direct contribution of MOG-IgG to epileptogensis is very unlikely, based on pathophysiological considerations and by findings that seizure occurrence did not differ between MOG-IgG positive (11%) and negative (14%) patients with ADEM, 34 one might hypothesize that subtle inflammation of brain cortex was already present at onset of seizures but not detectable on regular brain MRI. Indeed, all of these patients developed ADS or encephalitis symptoms and eventually MRI abormalities during follow-up. 119
Other possible disease presentations
Several cases of cranial neuritis with concurrent CNS involvement and MOG-IgG positivity have been described.120,121 The cranial nerve involvement was determined by contrast-enhancement on MRI at the nerve root entry zone. Possible explanations for MOG-IgG reactivity might include that at this anatomical region of the cranial nerve, the so called transitional zone, an overlap of central and peripheral myelin features exist, or that an inflammatory process from CNS lesions might progress down-stream. 120 Cases with isolated or a clear additional affection of the peripheral nervous system, such as cranial neuritis or inflammatory demyelinating polyneuropathy, remain elusive.122,123
MOG-IgG in daily clinical routine
Recently, a diagnostic algorithm for the inclusion of MOG-IgG in daily clinical practice in patients with ADS has been proposed, leading to four main phenotypes: MS, AQP4-positive NMOSD, MOG-IgG associated disorders and antibody-negative ADS (Figure 2).25,50,101,108 As MS is the most common ADS and frequently shows a characteristic MRI pattern as well as CSF-restricted oligoclonal bands, 101 it seems reasonable first to perform these two examinations. In the case of MS-atypical findings and negative AQP4-IgG, MOG-IgG should be determined. As the clinical phenotype associated with MOG-IgG expands to patients with autoimmune encephalitis, as shown by recent studies, MOG-IgG testing should also be considered in these cases especially after other differential diagnoses have been ruled out (Figure 2). It is crucial to limit MOG-IgG testing to these atypical cases, as screening of unselected, large populations for a rare biomarker generally decreases its positive predictive value by increasing the rate of false-positive results. Even if an assay shows a high specificity (e.g. ⩾99%), the true-positive results can easily be outnumbered by false-positive results if the prevalence of a biomarker is low and the number of samples tested is high. This fundamental statistical fact also applies to MOG-IgG testing. In order to avoid overdiagnosing MOG-IgG associated disorders, a list of indications as well as ‘red flags’ for MOG-IgG testing based on expert consensus have been recently proposed. 108

Spectrum of demyelinating diseases of the central nervous system.
Disappeareance of MOG-IgG is associated with a monophasic disease course
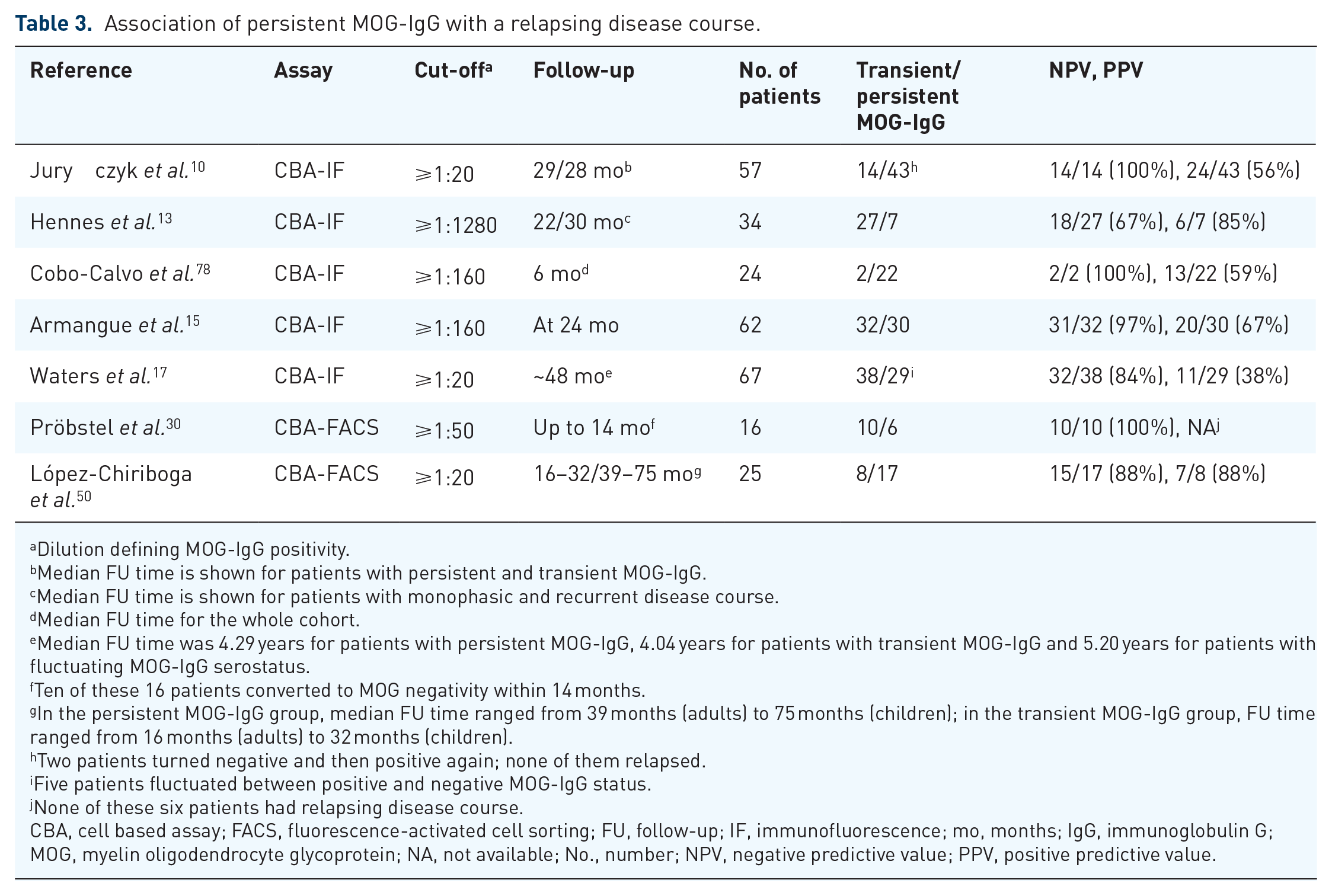
Approximately 35% of patients with MOG-IgG associated demyelinating disorders were reported to have a relapsing disease course, and relapes often manifest as optic neuritis irrespective of the initial type of disease manifestation.10,13,15–17,78 Most of the studies reported that a relapsing disease course was more likely in patients with higher MOG-IgG titres at onset13,78 and persisting MOG-IgG over time, whereas transient low titre MOG-IgG were typically associated with a monophasic disease course.10,13,15,17,30,37,46,50,78,124 Two recent studies each including more than 60 MOG-IgG positive patients with serial testing revealed that the median time to become seronegative was about 12 months.15,17 Overall, the predictive value of persisting MOG-IgG for the occurrence of relapses was only moderate (positive predictive value of approximately 60%); however, seroreversion to MOG-IgG negativity during early disease course reliably predicted monophasic disease (negative predictive value of approximately 90%). A summary of studies investigating the predictive value of MOG-IgG persistency is given in Table 3. Even though the predictive capability of MOG-IgG seems to be clear in general, there are still some limitations that do not allow their uncritical use in clinical routine and that have to be overcome first; for example, the usage of different cut-offs for defining antibody positivity due to various MOG-IgG assays, or the univariate statistical analyses that did not correct for the impact of other covariates, for example, immune treatment, on relapse risk and did not show the independent predictive value of MOG-IgG persistency. Furthermore, as the association of MOG-IgG with disease course and the calculation of its predictive value were determined by retrospective analyes only, studies that a priori apply, for example, a definition for MOG-IgG persistency and follow patients for a second attack are needed to clearly capture a clinically relevant predictive value of MOG-IgG.
Association of persistent MOG-IgG with a relapsing disease course.
Dilution defining MOG-IgG positivity.
Median FU time is shown for patients with persistent and transient MOG-IgG.
Median FU time is shown for patients with monophasic and recurrent disease course.
Median FU time for the whole cohort.
Median FU time was 4.29 years for patients with persistent MOG-IgG, 4.04 years for patients with transient MOG-IgG and 5.20 years for patients with fluctuating MOG-IgG serostatus.
Ten of these 16 patients converted to MOG negativity within 14 months.
In the persistent MOG-IgG group, median FU time ranged from 39 months (adults) to 75 months (children); in the transient MOG-IgG group, FU time ranged from 16 months (adults) to 32 months (children).
Two patients turned negative and then positive again; none of them relapsed.
Five patients fluctuated between positive and negative MOG-IgG status.
None of these six patients had relapsing disease course.
CBA, cell based assay; FACS, fluorescence-activated cell sorting; FU, follow-up; IF, immunofluorescence; mo, months; IgG, immunoglobulin G; MOG, myelin oligodendrocyte glycoprotein; NA, not available; No., number; NPV, negative predictive value; PPV, positive predictive value.
Treatment and outcome
Treatment of acute attacks
A significant proportion of patients with MOG-IgG associated disorders shows permament disability depending on the age of manifestation, with higher risk of disability in adults.10,13,15,78 In up to 60% of those patients, disability results from the onset attack, while in the remaining patients disability accumulation is due to the occurrence of further relapses.9,10,46,78 These findings imply that attention should be paid to acute management, because the time to treatment might be important for the prevention of permanent disability, as this is the case in NMOSD 125 and other types of autoimmune encephalitis. 126 Currently, there are no evidence-based guidelines for the acute treatment of patients with MOG-IgG associated disorders. Mostly, intravenous methylprednisolone and plasma exchange were used to treat acute attacks (Figure 3); however, intravenous immunoglobulins were also applied by some studies. 78 According to a seminal multicentre study that systematically investigated clinical and paraclinical features of patients with MOG-IgG associated disorders, intravenous methylprednisolone was applied at doses ranging from 1 to 2 g once a day for 3–5 days with good or complete recovery in approximately 50%. 9 If steroids did not result in recovery of symptoms, plasma exchange (usually with five cycles) was used as a second-line treatment, which further achieved substantial improvement in 40% of this steroid-non-responder. 9 Other studies reported even higher recovery rates from attacks of up to almost 90%; however, acute management was a mix of several consecutive treatments in a certain proportion of patients, that is, the effect with a single treatment was not assessed. 15
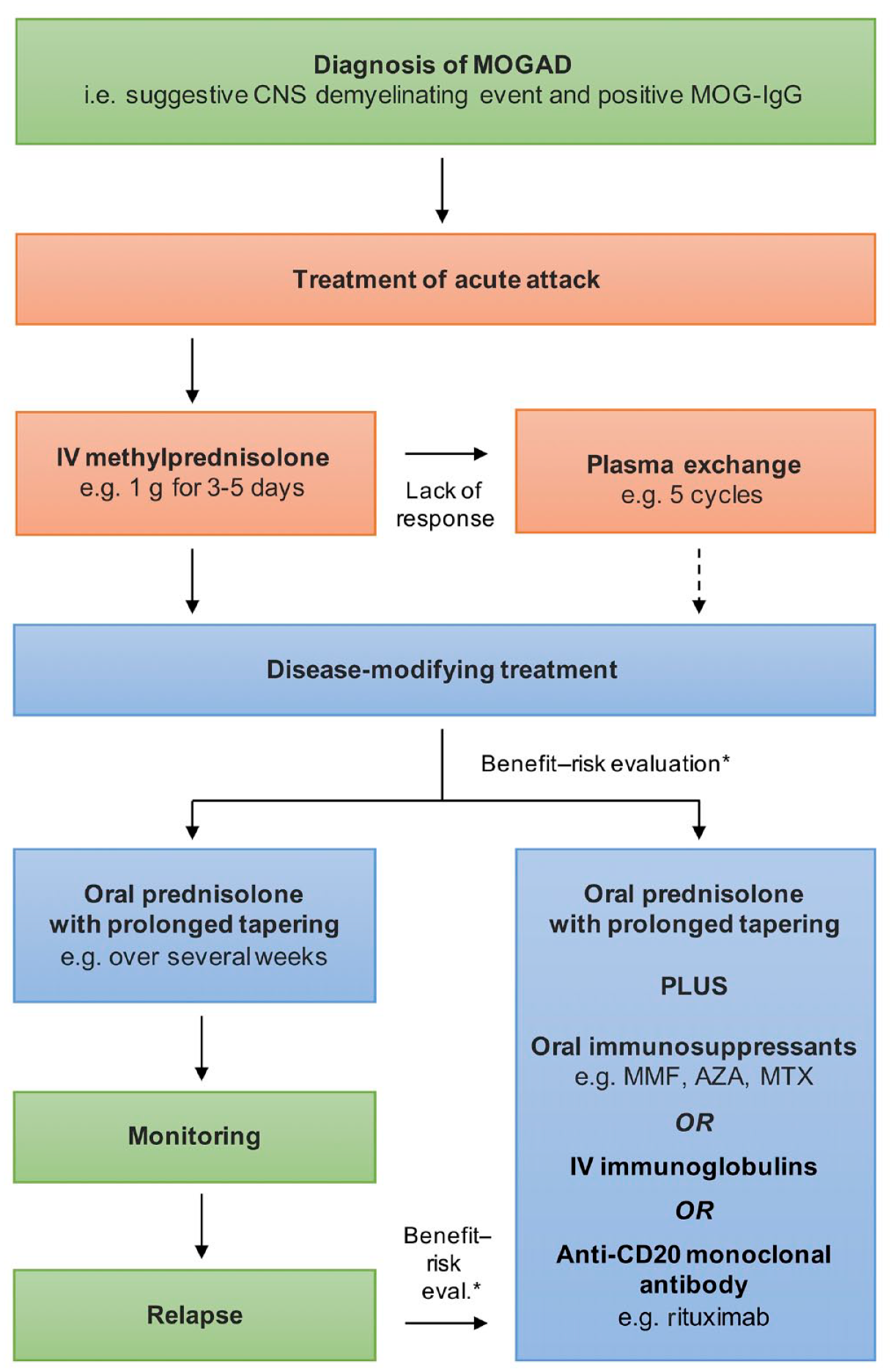
Therapeutic management of MOG-IgG associated disorders.
Disease-modifying treatment
Following treatment of the acute attack, a decision regarding the need for further disease-modifying treatment has to be made. Currently, there are no established parameters that allow a reliable risk evaluation for the occurrence of further relapses or disability. On these grounds, decision to start a preventive longer-lasting immune treatment remains to be made by an individual benefit–risk evaluation typically influenced, for example, by the severity of and the recovery from the acute attack, if more than one attack has occurred from the time to relapse and, if appropriate, from other factors that might be associated with the likelihood of relapse, for example, MOG-IgG persistency. 127 A treatment concept for patients with MOG-associated disorders is given in Figure 3.
Most immune treatments that are used in MOG-IgG associated disorders – which include corticosteroids, intravenous immunoglobulin, immunosuppressive drugs (such as mycophenolate mofetil, azathioprine and methotrexate) and rituximab – are associated with a reduction in time to relapse and annual relapse rate, respectively.9,10,78,84,85,128 Of note, immunomodulatory treatments for MS, such as interferon-β and glatiramer acetate, are ineffective.9,84 Most of the studies were retrospective, including various treatments, each with a small number of patients and, therefore, addressed only whether any treatment was superior to no treatment.
The main concrete conclusions drawn from these studies are described in the following. Relapses frequently occurred either during steroid weaning or shortly after its cessation.9,10,78,84,85 A recent study reported that relapses were mostly observed at doses <20 mg prednisone per day in adults or at doses <0.5 mg/kg per day in children. 85 The duration of treatment seems to impact relapse risk as well. The risk of relapse was higher in patients with only short-term immunosuppressive therapy (less than 3 months) as compared with those treated for a longer time. 10 Also of interest, one study showed that MOG-IgG positive patients treated with rituximab had some reduction of relapses, but relapse prevention was not associated with an effective depletion of memory B cells – in contrast to AQP4-positive NMOSD patients, in whom most relapses occurred after reemergence of memory B cells 129 and in whom B cell depleting therapies perform exceptionally well.130–132
However, a serious comparison of different therapies in terms of efficacy or even the estimation of the size of teatment effects is not feasible yet due to insufficient evidence (Class IV) and largely missing head to head analyses. Randomized controlled trials – as recently published for AQP4-positive NMOSD130–132 – are urgently needed also for MOG-IgG associated disorders. As such trials are difficult to perform due to the rarity of the disorder, profund statistical analyses adjusting for known covariates of real-world-data, for example, from national registries, are definitely an alternative. A recent prospective observational study showed an impressive risk reduction of relapse due to treatment with mycophenolate mofetil (MMF). 133 This study included 79 MOG-IgG positive patients presenting with ADEM, optic neuritis, transverse myelitis and/or brainstem syndrome compatible with deymelination who received either MMF or no immune treatment based on consensual decision with the treating physician and were followed for a median of 400 days; additional treatment for the acute phase with steroid tappering was allowed. Multivariate analyses adjusting for potential confounders such as age, sex, previous disease course and initial level of MOG-IgG titre revealed that MMF treatment resulted in a reduced risk of relapse by 86%. 133
Laboratory aspects
Validation of MOG-IgG assays
Only when measured using CBAs has an association between MOG-IgG and a non-MS demyelinating phenotype been established (reviewed by Reindl and Waters 2019 134 ). Recently, first blinded multicentre validation studies have addressed assay reproducibility between centres. The first study compared 394 samples using three different MOG-IgG CBAs from three international centres in a blinded validation experiment. 135 Overall, the concordance for all three CBAs was 98%, for the two live CBAs 99%. Clinical specificity ranged from 98.1% to 100%. Positive predictive values were higher for live CBAs (95.5% and 100%) than for fixed CBA (82.1%), whereas negative predictive values were comparable (78.8–79.8%). The second study compared three blinded samples in 13 centres using 13 different MOG-IgG CBAs. 136 Overall, the agreement of assays was 85%. Finally, the third study compared the reproducibility of 11 antibody assays for MOG-IgG [four live CBA-IF, three live CBA-FACS, one fixed CBA-IF and two enzyme-linked immunosorbent assay (ELISA)] and MOG-IgM (one live CBA-IF) from five international centres on 189 blinded samples. 137 Live MOG-IgG CBAs for samples previously identified as clearly positive or negative from four different national testing centres showed excellent agreement (96%) between the seven live CBAs for MOG-IgG. Agreement was lower with fixed CBA-IF (90%) and the ELISA showed no concordance with CBAs for detection of human MOG-IgG. All CBAs showed excellent inter-assay reproducibility. However, the agreement of MOG-IgG CBAs for borderline negative (77%) and particularly low positive (33%) samples was less good. Finally, most samples from healthy blood donors (97%) were negative for MOG-IgG in all CBAs.
In conclusion these three studies indicate that there is a good agreement of currently used live CBAs for high-titre, but not for low-titre, positive samples. We therefore recommend that results on the MOG-IgG status should include not only the qualitative results (i.e. positive or negative), but also a quantitative estimate (e.g. titre or FACS binding ratio with reference range) and the type of assay used. This would also help to clarify the presence of MOG-IgG in MS, which was re-assessed by two large studies. The first study analysed serum samples of 200 patients with chronic progressive MS and found that none of the patients was positive for MOG-IgG. 138 The second study analysed serum samples from 685 consecutive patients with MS, and found only two of them (0.3%) were MOG-IgG positive. 139 Both studies clearly indicate that MOG-IgG is rare in MS and if present indicate either insufficient assay specificity or an inappropriate clinical diagnosis.
The clinical relevance of CSF MOG-IgG
The clinical relevance of CSF MOG-IgG was recently re-analysed in 80 seronegative patients with demyelinating diseases (NMOSD and related diseases, MS). 140 Three seronegative cases (two NMOSD and one ADEM) had CSF MOG-IgG (4% of the whole cohort or 7% of cases excluding patients with MS). MOG-IgG were also detectable in the CSF of eight of 13 MOG-IgG seropositive cases, but in none of 36 patients with neurodegenerative disorders. This study and other previously published case reports reviewed by the authors indicate that analyzing CSF could improve diagnostic sensitivity in seronegative patients.
Pathology and pathophysiology of MOG-IgG associated diseases
The pathophysiology and neuropathology of autoimmune responses to MOG has been well established in animal models and has been reviewed in detail elsewhere. 6
So far, only case reports were available on the neuropathology associated with MOG-IgG in patients with inflammatory demyelinating diseases (reviewed by Reindl and Waters 134 ). The pathological features of MOG-IgG associated disorders were recently analysed in a larger series of two autopsies and 22 brain biopsies from patients with CNS inflammatory demyelinating diseases. 141 Both autopsies and the 22 brain biopsies had similar clinical, radiologic, laboratory and histopathological features. Pathology was dominated by the coexistence of both perivenous and confluent white matter demyelination, with an over-representation of intracortical demyelinated lesions compared with typical MS. Inflammatory cellular infiltrates were dominated by CD4+ T-cells and granulocytes. Complement deposition was present in all active white matter lesions, but a preferential loss of MOG was not observed. In contrast to the AQP4-IgG associated NMOSD pathology, AQP4 and astrocytes were preserved, whereas variable oligodendrocyte and axonal destruction was present. These results have most recently been confirmed by a second large case series from Japan. 142 Parallels with MOG-induced experimental autoimmune encephalomyelitis (EAE) suggest that MOG-IgG may be an amplification factor that augments CNS demyelination. Studies using the transfer of human MOG-IgG to experimental animal models indicated that human MOG-IgG can be pathogenic in rodents if they cross-react with rodent MOG and the titres and affinities of these antibodies are sufficiently high (reviewed by Reindl and Waters 134 ). Tissue injury is triggered by antibody-mediated injury or augmentation of inflammation caused by MOG-reactive T cells. When compared with AQP4-IgG the pathogenic role of human MOG-IgG is less evident. However, since the vast majority of MOG-IgG in patients are reactive only to human epitopes an appropriate test system available to determine their in vivo pathogenicity is still missing.
The importance of T-cell mediated inflammation was recently confirmed in two studies analysing serum and CSF cytokine and chemokine profiles in MOG-IgG positive patients.143,144 Both studies demonstrated that the CSF cytokine and chemokine profile associated with MOG-IgG is similar to AQP4-IgG positive NMOSD and distinct from MS. The inflammatory profile is characterized by coordinated upregulation of T helper 17 and other cytokines, particularly of interleukin-6.
Conclusion
In the last years, a multitude of studies using highly specific CBA have consolidated the clinical spectrum of ADS associated with MOG-IgG. Young children most often manifest with ADEM, whereas the typical clinical presentation of adults includes optic neuritis or myelitis [Figure 1(B)] with certain paraclinical features distinct from those observed in MS or AQP4-positive NMOSD (Table 2). Additional clinical phenotypes have also been described in MOG-IgG positive patients, for example, encephalitis other than ADEM 15 – frequently showing seizures and demyelinating events within intervals of months or years – as well as overlap syndromes with NMDAR encephalitis.
Apart from the important diagnostic value (Figure 2), MOG-IgG also imply some predictive capability. Early reversion of MOG-IgG to seronegativity shows a fair predictive value for a monophasic disease (Table 3), even though the predictive value of persisting MOG-IgG for a relapsing course is of minor importance. In clinical practice, one might conclude that MOG-IgG seroreversion encourages a ‘wait and see’ strategy, whereas the sole persistence of MOG-IgG does not justify uncritical initiation of long-term immune therapy.
Treatment of patients with MOG-IgG associated neurological disorders is still based on Class IV evidence (Figure 3); however, studies are now coming up that show at least controlled designs (with treatment and control arms) and, thus, will provide the urgently needed evidence of treatment efficacy. In the light of all this amazing clinical progress, high-quality and specific MOG-IgG assays are – more than ever – of utmost importance.
Footnotes
Conflict of interest statement
The University Hospital and Medical University of Innsbruck (Austria; employer of HH and MR) receives payments for antibody assays (MOG, AQP4 and other autoantibodies) and for MOG and AQP4 antibody validation experiments organized by Euroimmun (Lübeck, Germany).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Markus Reindl is supported by a research grant from the Austrian Science Fund (FWF, project P32699).