
Abstract
Background:
In the phase III eculizumab for refractory generalized myasthenia gravis REGAIN study [ClinicalTrials.gov identifier: NCT01997229] and its open-label extension (OLE) [ClinicalTrials.gov identifier: NCT02301624], patients with treatment-refractory antiacetylcholine receptor antibody-positive generalized myasthenia gravis had clinically meaningful improvements with eculizumab versus placebo. This subgroup analysis evaluated data from patients with a recent history of chronic intravenous immunoglobulin (IVIg) use before study entry.
Methods:
The subgroup comprised patients who had received IVIg at least four times in 1 year, with at least one IVIg treatment cycle during the 6 months before the first REGAIN study dose. Data from REGAIN and the OLE were analyzed. Response to eculizumab versus placebo was assessed using four validated, disease-specific measures. Incidences of exacerbations and safety endpoints were recorded.
Results:
The subgroup had similar patient and disease characteristics as the overall REGAIN population. Clinical assessments showed sustained eculizumab efficacy during REGAIN and the OLE over 18 months. Patients receiving placebo in REGAIN experienced rapid improvements in assessment scores when treated with eculizumab in the OLE. There was a lower rate of disease exacerbations with eculizumab than with placebo during REGAIN, and eculizumab was well tolerated.
Conclusion:
Eculizumab treatment, compared with placebo, results in meaningful clinical improvements and fewer disease exacerbations for patients who previously received chronic IVIg.
Trial registration:
REGAIN [ClinicalTrials.gov identifier: NCT01997229]; REGAIN open-label extension [ClinicalTrials.gov identifier: NCT02301624].
Introduction
Approximately 15% of patients with generalized myasthenia gravis (gMG) do not respond to standard immunosuppressive therapies (ISTs), or require intravenous immunoglobulin (IVIg) or plasma exchange (PLEX) to manage their symptoms.1–3 For these patients with treatment-refractory myasthenia gravis (MG), disease control is impaired; a study in two US health-plan databases reported 4-fold and 4.7-fold increases in rates of MG exacerbations/crises, in refractory versus nonrefractory cases, respectively. 4 A UK study reported increased healthcare resource use in refractory versus nonrefractory MG. 5 It is important, therefore, to consider therapeutic needs in treatment-refractory MG.
IVIg is a proven short-term therapy for MG exacerbations/crises, and is also considered as a maintenance therapy in treatment-refractory MG inadequately controlled with standard IST. 3 The longevity of its effects is limited, however, and there are some tolerability issues associated with chronic IVIg therapy.4,6 A well-tolerated treatment option with sustained effectiveness is therefore needed for these patients to minimize the risk of MG exacerbations/crises.
Differences in treatment responsiveness in gMG may reflect underlying biological differences in disease pathogenesis. 2 Among the multiple targets of IVIg in the immune regulatory network is the complement system, 7 which mediates neuromuscular junction damage in gMG. This suggests that patients with gMG who require chronic IVIg for symptom management may respond to therapies that specifically inhibit the complement cascade.
The 6-month, phase III, randomized, double-blind, placebo-controlled eculizumab for refractory generalized myasthenia gravis REGAIN study [ClinicalTrials.gov identifier: NCT01997229] evaluated the terminal complement inhibitor eculizumab in patients with antiacetylcholine receptor antibody-positive (AChR+) gMG failing to achieve symptom control despite recent treatment with at least two ISTs, or one IST and chronic IVIg or PLEX. 8 REGAIN demonstrated improvements in MG Activities of Daily Living (MG-ADL) and Quantitative MG (QMG) scores from baseline to week 26. MG exacerbation rates and rescue therapy use were lower with eculizumab than with placebo, and eculizumab was well tolerated. Patients completing REGAIN were eligible to enter the open-label extension (OLE) study [ClinicalTrials.gov identifier: NCT02301624], which was completed in January 2019 (last patient, last visit). Data from the OLE demonstrated long-term safety and sustained efficacy of eculizumab. 9 We report a subgroup analysis from REGAIN and its OLE to evaluate the response to eculizumab over a period of up to 18 months in patients receiving chronic IVIg before participating in REGAIN.
Methods
Patient consent and ethical approval
All patients provided written, informed consent for participation in both trials. Independent ethics committees or institutional review boards provided written approval for the study protocol and all study amendments (supplemental material). The studies were performed in accordance with the ethical standard laid down in the 1964 Declaration of Helsinki and are registered with www.clinicaltrials.gov [ClinicalTrials.gov identifiers: NCT01997229 and NCT02301624 for REGAIN and the REGAIN OLE, respectively].
Study design
The methodology for REGAIN and the OLE has been published.8,9 Briefly, eligible patients were randomized (1:1) to eculizumab (induction dosing 900 mg on day 1 and weeks 1, 2 and 3; 1200 mg at week 4; then maintenance dosing 1200 mg every second week) or placebo on the same schedule. 8 Participants from both groups completing REGAIN could enter the OLE, during which all patients received 1200 mg open-label eculizumab every 2 weeks after a 4-week blinded induction protocol. 9 The final OLE database was locked on 8 March 2019.
Study population
Data were evaluated from patients receiving IVIg before REGAIN at least four times in 1 year, with at least one IVIg treatment administered within 6 months before the first dose of REGAIN study drug. Patients were excluded from REGAIN if they had received IVIg in the 4 weeks before randomization, and concomitant IVIg use was only permitted during REGAIN/OLE as rescue therapy.8,9
Assessments
Response was assessed using four validated, disease-specific measures [MG-ADL, QMG, MG Composite scale (MGC) and the 15-item MG Quality of Life Questionnaire (MG-QOL15)]. A clinical response was defined as at least a 3-point improvement in MG-ADL total score or at least a 5-point improvement in QMG total score at the evaluation time point compared with REGAIN baseline. The incidence of exacerbations (defined as an MG crisis, significant symptomatic worsening or requirement for rescue therapy) was evaluated throughout REGAIN/OLE, and during the year before REGAIN enrollment. Safety endpoints were assessed throughout REGAIN/OLE.
Statistical analysis
Changes from REGAIN baseline in MG-ADL, QMG, MGC and MG-QOL15 mean total scores were based on t tests. Data are presented as the mean change from REGAIN baseline; mean differences between treatment groups are presented as 95% confidence intervals (CIs). Subgroup analyses were not powered to detect significant differences owing to the small sample size; hence, p values are not presented. The responder analyses measured the proportion of patients with clinically meaningful improvements from REGAIN baseline. Exact (Clopper–Pearson) 95% CIs for the responder proportions are presented. For exacerbations, model-based event rates per 100 patient-years were calculated following previously published methodology. 9 All statistical analyses were performed using SAS version 9.4 (SAS Instittute, Cary, NC, USA).
Results
Patients
In total, 18 patients were included (eculizumab, n = 9; placebo, n = 9). One eculizumab-treated patient withdrew from REGAIN (to receive IVIg after experiencing MG clinical deterioration); 17 patients continued into the OLE (eculizumab/eculizumab, n = 8; placebo/eculizumab, n = 9).
Baseline demographics and characteristics, including mean baseline scores for MG-ADL, QMG, MGC and MG-QOL15, were generally similar for both subgroup treatment arms and the overall REGAIN population. 8 The duration between last recorded IVIg dose end date and first REGAIN study dose was 4–16 weeks (n = 17; 1 patient missing accurate end-date data). In the year before entering REGAIN, 9/18 patients experienced at least one exacerbation (eculizumab: 6 events in 4 patients; placebo: 19 events in 5 patients); there was also 1 patient with MG crisis in the eculizumab group. Compared with the overall REGAIN population, 9 this subgroup experienced a higher rate of exacerbations in the year before REGAIN start (150.0 versus 102.4 exacerbations/100 patient-years).
Clinical response
At REGAIN week 26, eculizumab-treated patients had numerically larger improvements from baseline in MG-ADL and QMG mean total scores than placebo-treated patients (Table 1). Improvements from baseline in MG-ADL, QMG, MGC and MG-QOL15 mean total scores with eculizumab during REGAIN were sustained in the eculizumab/eculizumab group during the OLE. Patients receiving open-label eculizumab after placebo during REGAIN experienced rapid improvements in assessment scores.
Change from REGAIN baseline in MG-ADL, QMG, MGC and MG-QOL15 mean total scores to REGAIN week 26 and open-label extension weeks 26 and 52 in patients with gMG who had previously used chronic intravenous immunoglobulin.
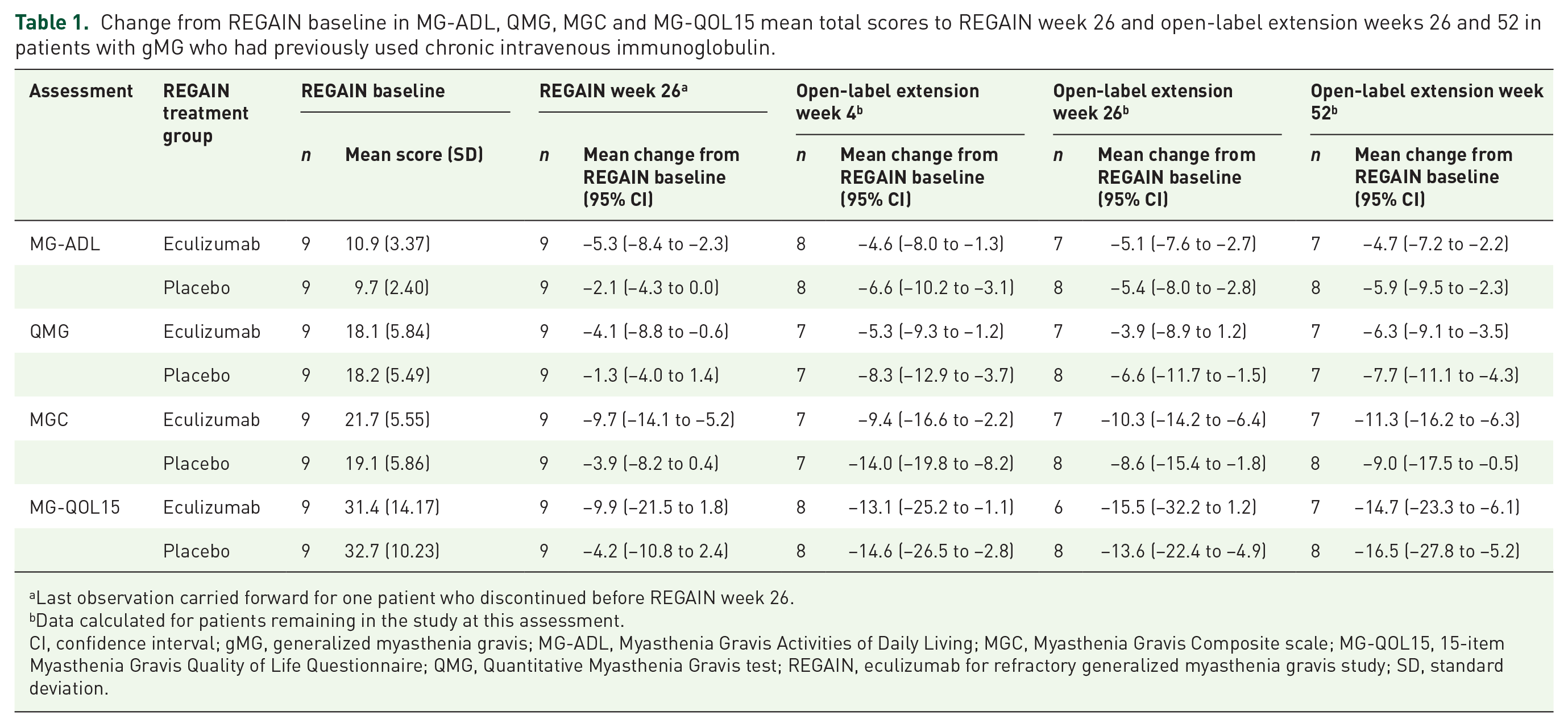
Last observation carried forward for one patient who discontinued before REGAIN week 26.
Data calculated for patients remaining in the study at this assessment.
CI, confidence interval; gMG, generalized myasthenia gravis; MG-ADL, Myasthenia Gravis Activities of Daily Living; MGC, Myasthenia Gravis Composite scale; MG-QOL15, 15-item Myasthenia Gravis Quality of Life Questionnaire; QMG, Quantitative Myasthenia Gravis test; REGAIN, eculizumab for refractory generalized myasthenia gravis study; SD, standard deviation.
A clinical response (MG-ADL or QMG) was achieved by most subgroup patients receiving eculizumab during REGAIN and the OLE (Figure 1). In contrast, only a third or fewer subgroup patients achieved a clinical response while receiving placebo in REGAIN (Figure 1). Most of these patients subsequently achieved a clinical response (MG-ADL or QMG) after receiving eculizumab during the OLE.

Clinical response in REGAIN and the open-label extension study, versus REGAIN baseline.
Exacerbations
During REGAIN, 4/18 patients experienced exacerbations; 1 eculizumab-treated patient and 3 placebo-treated patients experienced 7 and 12 exacerbations, respectively. During the OLE, 6/17 patients experienced exacerbations; all 6 were in the eculizumab/eculizumab group. Most exacerbations occurring in eculizumab-treated patients during REGAIN (7/7 exacerbations) and the OLE (9/15 exacerbations) were experienced by 1 patient. The exacerbation rate was reduced by 68.6% from 150.0 exacerbations/100 patient-years in the year before REGAIN (n = 18) to 47.0 exacerbations/100 patient-years during eculizumab treatment in REGAIN and the OLE (n = 17; p = 0.1531). This exacerbation rate in the OLE population (n = 17) also compared favorably with that in the REGAIN placebo group (n = 9; 240.9 exacerbations/100 patient-years; 80.4% reduction, p = 0.059).
Safety
The safety profile of eculizumab in the chronic IVIg subgroup was consistent with that in all eculizumab-treated patients in REGAIN.8,9 The most common adverse events with eculizumab in this subgroup were headache (REGAIN, 22.2%; OLE, 52.9%; versus REGAIN placebo, 11.1%) and upper respiratory tract infection (33.3%, 47.1% and 33.3%, respectively). One patient in this subgroup analysis died during the OLE. This death was attributed to end-stage liver disease (considered by the investigator unlikely to be related to the use of eculizumab); the patient had cryptogenic liver cirrhosis and a history of fatty liver.
Discussion
Patients with treatment-refractory gMG may be treated with chronic IVIg or PLEX. 3 Among the overall REGAIN population, 18 patients received chronic IVIg in the 6 months before study entry. This subanalysis assessed the patients in REGAIN with a recent history of chronic IVIg treatment.
Although baseline patient characteristics were similar to those in the REGAIN population, this subgroup had previously demonstrated poorly controlled disease by virtue of the need for chronic IVIg and the high exacerbation rate in the year before the start of the study. 9 The benefits and tolerability of eculizumab demonstrated in REGAIN supported its approval in Europe and Japan for adults with treatment-refractory AChR+ gMG, and in the USA for adults with AChR+ gMG.
During the 26-week REGAIN study and the OLE, eculizumab was associated with sustained improvements from baseline in all assessment scores in the chronic IVIg subgroup. As in the overall OLE population, 9 subgroup patients who received placebo in REGAIN experienced a rapid and sustained clinical response with eculizumab. During REGAIN, MG exacerbations were more frequent in subgroup patients receiving placebo versus eculizumab, consistent with active disease in this cohort. The response to the terminal complement inhibitor eculizumab in patients who were refractory to other ISTs and who had previously required chronic IVIg therapy, which acts at least in part by diminishing complement activity, 7 strengthens the evidence for the role of complement inhibition in the effective treatment of gMG. With its favorable safety profile in previous chronic IVIg users, which was similar to that in the overall study population, and its sustained effects in these patients, eculizumab may constitute a valuable treatment option for patients who require chronic IVIg for symptom management.
There were 15 exacerbations in the eculizumab/eculizumab group during the OLE, but none in patients receiving placebo in REGAIN and then eculizumab (placebo/eculizumab). This discrepancy is largely attributable to one patient, who was the only eculizumab-treated patient in the subgroup to experience any exacerbations during REGAIN and who contributed over half of all exacerbations reported in the OLE. The impact of a single patient’s data on overall study findings highlights the limitation of the small sample size. The exacerbation rate during eculizumab treatment in REGAIN and the OLE was reduced considerably from that in the year before REGAIN start and from that in the REGAIN placebo group, yet these differences did not reach statistical significance, probably owing to inadequate power with the small sample size.
Selection bias in the OLE population is unlikely because over 90% of both REGAIN participants (117/125) and chronic IVIg subgroup members (17/18) enrolled in the OLE. It is possible, however, that some selection bias may have been introduced in the REGAIN population as a result of the requirement to have an IVIg washout period of 4 weeks before randomization. Interestingly, in a recent case series, 13 patients with refractory gMG receiving maintenance IVIg therapy transitioned to eculizumab after a shorter IVIg washout period of 10–14 days without significant safety concerns and with clinically meaningful improvements in outcomes. 10
Conclusion
For patients who had previously received chronic IVIg, eculizumab resulted in rapid improvements in MG signs and symptoms across four validated measures of disease severity, which were maintained over 18 months. There were also fewer MG exacerbations with eculizumab compared with placebo during REGAIN. Eculizumab may therefore provide long-term benefits for patients with AChR+ gMG who would otherwise require chronic IVIg treatment.
Supplemental Material
sj-pdf-1-tan-10.1177_1756286420911784 – Supplemental material for Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study
Supplemental material, sj-pdf-1-tan-10.1177_1756286420911784 for Response to eculizumab in patients with myasthenia gravis recently treated with chronic IVIg: a subgroup analysis of REGAIN and its open-label extension study by Saiju Jacob, Hiroyuki Murai, Kimiaki Utsugisawa, Richard J. Nowak, Heinz Wiendl, Kenji P. Fujita, Fanny O’Brien and James F. Howard in Therapeutic Advances in Neurological Disorders
Footnotes
Acknowledgements
The authors would like to thank the patients who took part in REGAIN and the OLE study, as well as their families. Completion of such a large, multinational study could not have been accomplished without the participation of our investigators (a list of the investigators associated with the patients included in this subgroup analysis is provided below and in the supplemental material). We thank them for their contributions to the completion of the study. We also thank Sivani Paskaradevan (Alexion Pharmaceuticals) for critical review of the manuscript and Cindy Lane (formerly of Alexion Pharmaceuticals) for clinical study oversight. We would like to acknowledge Vicky Sanders, PhD, of Oxford PharmaGenesis, Oxford, UK, who provided medical writing support in the production of this manuscript (funded by Alexion Pharmaceuticals).
Authors’ note
Data included in this manuscript have been published as congress abstracts and presented as posters for the American Association of Neuromuscular and Electrodiagnostic Medicine Annual Meeting (10–13 October 2018, Washington, DC, USA) and at the 15th International Congress on Neuromuscular Diseases (6–10 July 2018, Vienna, Austria).
Author contributions
KPF, FOB and JFH contributed to the concept/design of the study; all authors contributed to data acquisition, analysis or interpretation, drafting/critical revision and final approval of the manuscript.
REGAIN study subgroup investigators
Claudio Gabriel Mazia, Miguel Wilken, Fabio Barroso, Juliet Saba, Jan De Bleecker, Guy Van den Abeele, Kathy de Koning, Katrien De Mey, Alzira Alves de Siqueira Carvalho, Igor Dias Brockhausen, David Feder, Daniel Ambrosio, Pamela César, Ana Paula Melo, Renata Martins Ribeiro, Rosana Rocha, Bruno Bezerra Rosa, Thabata Veiga, Luiz Augusto da Silva, Murilo Santos Engel, Jordana Gonçalves Geraldo, Kimiaki Utsugisawa, Yuriko Nagane, Ikuko Kamegamori, Tomoko Tsuda, Yuko Fujii, Kazumi Futono, Yukiko Ozawa, Aya Mizugami, Yuka Saito, Anneke van der Kooi, Marianne de Visser, Tamar Gibson, Seung Min Kim, Ha-Neul Jeong, JinWoo Jung, Yool-hee Kim, Hyung Seok Lee, Ha Young Shin, Eun Bi Hwang, Miju Shin, Josep Gamez Carbonell, Pilar Sune, Maria Salvado Figueras, Gisela Gili, Gonzalo Mazuela, Fredrik Piehl, Albert Hietala, Lena Bjarbo, Sevim Erdem- Ozdamar, Can Ebru Bekircan-Kurt, Nazire Pinar Acar, Ezgi Yilmaz, Yagmur Caliskan, Gulsah Orsel, Anthony Amato, Thomas Cochrane, Mohammed Salajegheh, Kristen Roe, Katherine Amato, Shirli Toska, Jonathan McKinnon, Laura Haar, Naya McKinnon, Karan Alcon, Kaitlyn McKenna, Nadia Sattar, Kevin Daniels, Dennis Jeffery, Tahseen Mozaffar, Tiyonnoh Cash, Namita Goyal, Gulmohor Roy, Veena Mathew, Fatima Maqsood, Brian Minton, Charlene Hafer-Macko, Justin Kwan, Lindsay Zilliox, Karen Callison, Valerie Young, Beth DiSanzo, Kerry Naunton, Tuan Vu, Lara Katzin, Terry McClain, Brittany Harvey, Adam Hart, Kristin Huynh, Said Beydoun, Amaiak Chilingaryan, Victor Doan, Brian Droker, Hui Gong, Sanaz Karimi, Frank Lin, Terry McClain, Krishna Polaka, Akshay Shah, Anh Tran, Salma Akhter, Ali Malekniazi, Rup Tandan, Michael Hehir, Waqar Waheed, Shannon Lucy, Tulio Bertorini, Thomas Arnold, Kendrick Hendersen, Rekha Pillai, Ye Liu, Lauren Wheeler, Jasmine Hewlett, Mollie Vanderhook
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was funded by Alexion Pharmaceuticals.
Conflict of interest statement
Saiju Jacob has served as a paid consultant for Alnylam Pharmaceuticals and Alexion Pharmaceuticals, and has received speaker honoraria from Bio Products Laboratory, UK, Eisai Ltd and Terumo BCT.
Hiroyuki Murai has served as a paid consultant for Alexion Pharmaceuticals, argenx and Ra Pharma, and has received speaker honoraria from the Japan Blood Products Organization and research support from the Ministry of Health, Labor and Welfare, Japan.
Kimiaki Utsugisawa has served as a paid consultant for Alexion Pharmaceuticals, argenx, Ra Pharma and UCB Pharma.
Richard Nowak has received research support from Alexion Pharmaceuticals, Genentech, Grifols, Momenta, the Myasthenia Gravis Foundation of America, the National Institutes of Health (including the National Institute of Allergy and Infectious Diseases and the National Institute of Neurological Disorders and Stroke) and Ra Pharma, and has served as a paid consultant for Alexion Pharmaceuticals, CSL Behring, Grifols, Momenta, Ra Pharma, Roivant Sciences and Shire, a Takeda company.
Heinz Wiendl received honoraria for participation in scientific advisory boards and consultation for Biogen, Evgen Pharma, MedDay Pharmaceuticals, Merck Serono, Novartis, Roche Pharma AG and Sanofi-Genzyme, and speaker honoraria and travel support from Alexion Pharmaceuticals, Biogen, Cognomed, F. Hoffmann-La Roche Ltd, Gemeinnützige Hertie-Stiftung, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, Teva and WebMD Global. He has been a paid consultant for AbbVie, Actelion, Biogen, IGES, Novartis, Roche Pharma AG, Sanofi-Genzyme and the Swiss Multiple Sclerosis Society. His research is funded by Biogen GmbH, Deutsche Forschungsgesellschaft (DFG), Else Kröner-Fresenius Foundation, the German Federal Ministry of Education and Research (BMBF), GlaxoSmithKline GmbH, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Münster, NRW Ministry of Education and Research, RE Children’s Foundation, Roche Pharma AG and Sanofi-Genzyme.
Kenji Fujita was an employee of, and owns stock in, Alexion Pharmaceuticals and is employed by Alnylam Pharmaceuticals.
Fanny O’Brien is an employee of, and owns stock in, Alexion Pharmaceuticals.
James Howard Jr reports research support from Alexion Pharmaceuticals, argenx, the Centers for Disease Control and Prevention (Atlanta, GA, USA), the Muscular Dystrophy Association, the National Institutes of Health (including the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Institute of Neurological Disorders and Stroke) and Ra Pharma; he has received consulting fees from Alexion Pharmaceuticals, argenx, Ra Pharma and VielaBio; and nonfinancial support from Alexion Pharmaceuticals, argenx and Ra Pharma.
Data-sharing statement
Qualified academic investigators may request participant-level, de-identified clinical data and supporting documents (statistical analysis plan and protocol) pertaining to this study. Further details regarding data availability, instructions for requesting information and our data disclosure policy are available on the Alexion.com website (![]() ).
).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.