
Abstract
Background:
PF-06649751 is a novel, oral, non-catechol-based, D1/D5 dopamine receptor partial agonist under investigation for the treatment of motor symptoms associated with Parkinson’s disease.
Methods:
A 15-week, phase II, double-blind, placebo-controlled clinical trial was conducted to assess the efficacy and safety of flexible-dose PF-06649751 in subjects with early stage Parkinson’s disease (ClinicalTrials.gov identifier: NCT02847650).
Results:
Enrollment was terminated early for reasons unrelated to the trial. Overall, 57 subjects received study medication (PF-06649751 = 29; placebo = 28) and 47 completed the study (PF-06649751 = 25; placebo = 22). Despite early termination, the study met its primary endpoint with the PF-06649751 group showing statistically significant improvement from baseline in the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III score at week 15 compared with placebo. Mean (SE) change in MDS-UPDRS Part III score was −9.0 (1.54) for PF-06649751 and −4.3 (1.65) for placebo. This corresponds to an improvement versus placebo of 4.8 for the PF-06649751 group (two-sided p = 0.0407; 90% CI = 1.0, 8.6). Statistically significant improvement in MDS-UPDRS-III score was also observed at all assessment time points prior to week 15. The safety profile of PF-06649751 was similar to that observed in prior studies, with the majority of adverse events (AEs) reported as mild or moderate. The most common AEs in the PF-06649751 group were nausea, headache, dry mouth, somnolence, and tremor.
Conclusions:
Once-daily dosing of oral PF-06649751 resulted in significant improvement of motor symptoms and was generally well tolerated in subjects with early stage Parkinson’s disease.
Introduction
Loss of dopaminergic neurons underlies the hallmark motor symptoms of Parkinson’s disease.1–5 Standard treatment remains the restoration of dopaminergic function through administration of the synthetic dopamine precursor levodopa (
There has long been interest in the potential of D1- or D2-selective receptor agonists as an alternative to levodopa in subjects with Parkinson’s disease.13–15 Clinical trials of limited duration with prototype full D1 agonists demonstrated that, while these agents exhibited robust anti-parkinsonian effects on motor symptoms, challenges with pharmacokinetics, cardiovascular parameters, and side effects including dyskinesias made them ill-suited for further clinical advancement.16–18 In contrast, efforts to develop D2-selective agonists with suitable clinical properties were more successful, with several receiving approval for the treatment of Parkinson’s disease, including pramipexole, ropinirole, and rotigotine.19–21 In general, these agents exhibit less-frequent motor complications than levodopa, but are not as effective for controlling motor symptoms.2,9,22 D2-selective agonists are also associated with an increased incidence of specific adverse effects (AEs), including nausea, somnolence, hypotension, compulsive behaviors, and hallucinations, particularly in the elderly.19–21,23 Thus, there remains a need for agents that provide effective and predictable control of motor symptoms while avoiding side effects associated with activation of D2-like receptors. Existing clinical and preclinical data provide theoretical support for the long-standing hypothesis that selective activation of D1-like receptors may be an important therapeutic option for Parkinson’s disease.13–15 To date, however, attempts to develop D1-selective agonists have been hampered by tolerability issues (notably, acute hypotension) and poor pharmacokinetics (e.g. short half-life and low oral availability). 15
PF-06649751 is a potent, highly selective, orally administered, dopamine D1/D5 receptor partial agonist being evaluated for the once-daily symptomatic treatment of Parkinson’s disease. It is part of a new class of novel non-catechol-based agents designed to avoid the pharmacokinetic and tolerability concerns observed with previously discontinued D1 agonists. 24 PF-06649751 has demonstrated preliminary evidence of safety and efficacy in two short-term phase I clinical trials in subjects with idiopathic Parkinson’s disease, based on the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III scores. 24 Here, we report findings from a phase II study examining the efficacy, safety, and tolerability of once-daily PF-06649751 in subjects with early stage Parkinson’s disease in a 15-week outpatient setting.
Methods
Study design
This phase II, double-blind, randomized, placebo-controlled, flexible-dose, 15-week study in subjects with early stage Parkinson’s disease was conducted from October 2016 (first subject first visit) to January 2018 (last subject last visit) at 23 sites in the United States, France, Germany, and Israel (ClinicalTrials.gov identifier: NCT02847650). The study protocol was approved by the appropriate institutional review board or independent ethics committee at each participating investigational center, and all subjects provided written informed consent prior to entering the study. This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines.
The study consisted of a 30-day screening period, a 15-week double-blind treatment period (9 week dose optimization and 6 week dose maintenance), and a 28-day follow-up period. The schedule of planned study visits can be seen in Figure 1
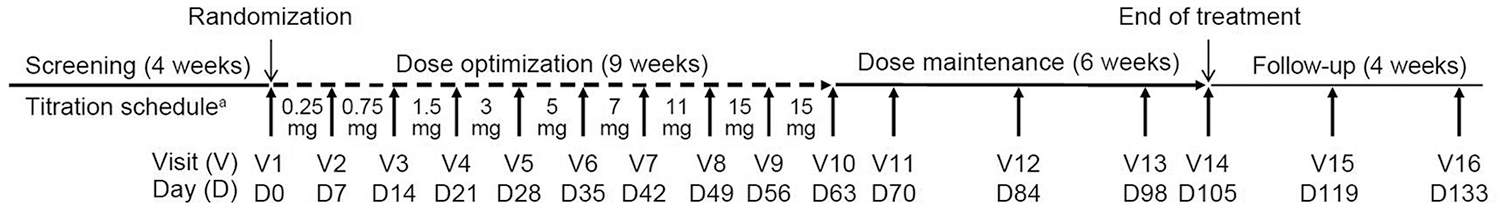
Design of the study.
Dosing
At Visit 1, subjects were stratified by region and randomized (1:1) in a double-blind manner to either oral (tablet) PF-06649751 or matching placebo. Subjects administered medication once-daily (morning) and recorded administration in a dosing diary. Compliance was assessed at each visit based on review of medication blister packs and the subject’s dosing diary. The recommended dosing schedule was as follows: 0.25 mg (days 1–7), 0.75 mg (days 8–14), 1.5 mg (days 15–21), 3 mg (days 22–28), 5 mg (days 29–35), 7 mg (days 36–42), 11 mg (days 43–49), and 15 mg (from day 50 onward). A dose increase from 0.25 mg to 0.75 mg was mandatory at Visit 2, and subjects who could not tolerate 0.25 mg were discontinued. Dosing decisions (increase/decrease/stay at current dose) were made on a weekly basis thereafter, up to Visit 10, based on investigator assessment of an individual subject achieving an optimal balance of motor symptom control and tolerability. Dosing adjustments after week 10 required consultation with the sponsor’s study clinician. The minimum recommended dose level was 3 mg.
Inclusion and exclusion criteria
Study inclusion criteria were as follows: males agreeing to use contraception or females of nonchildbearing potential; aged 45–80; a clinical diagnosis of Parkinson’s disease (Hoehn & Yahr Stage I–III); an MDS-UPDRS Part III score ⩾10; treatment naïve or history of prior incidental treatment with dopaminergic agents for no more than 28 days and not within at least 7 days prior to randomization; a Mini-Mental State Examination (MMSE) score ⩾26; body mass index (BMI) of 17.5–35 kg/m2; and a total body weight ⩾45 kg.
Key exclusion criteria included: history or clinical features consistent with an atypical Parkinsonian syndrome (e.g. progressive supranuclear palsy, multiple system atrophy, and cortico basal degeneration); significant psychiatric disease (except minor or treated stable depression); risk of suicide; current malignancy (except nonmetastatic basal or squamous cell carcinoma of the skin); significant cardiovascular disease; cognitive impairment that would interfere with ability to comply with study procedures; pregnancy; currently breastfeeding; male subjects with pregnant partner; 12 lead electrocardiogram (ECG) demonstrating a QTcF interval >450 ms (>470 ms for females) or a QRS interval >120 ms.
Concomitant medications
Stable doses of antihypertensives, antidepressants [other than irreversible monoamine oxidase (MAO) A/B inhibitors], anticoagulants, lipid lowering agents, oral antidiabetics, thyroid replacement hormones, antiemetics, antacids, and opioids (low doses for chronic pain) were permitted throughout the study. MAO B inhibitors, amantadine, and anticholinergic drugs were permitted if subjects were on stable doses of these anti-parkinsonian medications for at least 42 days prior to Visit 1.
Prior treatment with levodopa or dopamine receptor agonists for more than 28 days, or any treatment with these agents within 7 days of Visit 1, was prohibited. Rescue anti-parkinsonian medication use after first dose of study treatment was not permitted. Marijuana and grapefruit (and related citrus fruits/juice) were prohibited throughout the study, and subjects abstained from alcohol for at least 12 h prior to study visits. Stimulants, antipsychotics, metoclopramide, reserpine, lithium, tricyclic antidepressants, irreversible MAO A/B inhibitors, and anti-epileptics (except gabapentin or pregabalin for chronic pain) were not permitted within 42 days of Visit 1 and throughout the study.
Primary efficacy measure
The primary study endpoint was the change, from baseline, in MDS-UPDRS Part III score at week 15. 25 MDS-UPDRS Part III scores were also compared between treatment groups at weeks 3, 5, 7, 9, and 12.
Safety and tolerability measures
Safety and tolerability measures included AE reporting, clinical laboratory parameters, vital signs, ECG parameters, the Beck Depression Inventory-II (BDI-II), Epworth Sleepiness Scale (ESS), the Columbia Suicide Severity Rating Scale (C-SSRS), the Questionnaire for Impulsive Compulsive Disorders in Parkinson’s Disease-Rating Scale (QUIP-RS), and the Physician Withdrawal Checklist (PWC-20).26–30
Exploratory endpoints
Change, from baseline, in exploratory endpoint measures was assessed at weeks 9 and 15, and included Patient Global Impression-Severity (PGI-S), Patient Global Impression-Improvement (PGI-I), Parkinson’s Disease Questionnaire (PDQ-39), EuroQol 5 Dimension (EQ-5D-5L), Oral Symbol Digit Modality Test (OSDMT), Modified Clinical Global Impression-Severity (MCGI-S), Modified Clinical Global Impression-Improvement (MCGI-I), and MDS-UPDRS total and component scores.31,32
Statistical analysis
Sample size was based on the primary study endpoint of change, from baseline, in MDS-UPDRS Part III score at week 15 for PF-06649751 versus placebo. The decision criteria for efficacy were: (1) a ⩾50% confidence that the PF-06649751 effect is at least 3.6 points better than placebo (i.e. a mean improvement of at least 3.6 points over placebo), and (2) a ⩾95% confidence that the PF-06649751 effect over placebo is greater than 0. Note that a decrease in MDS-UPDRS Part III score represents improvement in motor function. It was estimated that 34 subjects per arm would give sufficient precision to meet both criteria, and, assuming that 23% of subjects would fail to complete the study or optimize the dose, 44 subjects per arm were planned.
All efficacy analyses were based on the Full Analysis Set, defined as all subjects who received at least one dose of study medication and had baseline and at least one post-baseline MDS-UPDRS Part III score. Most endpoints (see exceptions below) were analyzed using a restricted maximum likelihood-based mixed model for repeated measures that included change from baseline to each postbaseline visit as the response variable and fixed effects of treatment, visit, treatment-by-visit interaction, baseline score, baseline-by-visit interaction, geographic region, and concurrent anti-Parkinson’s disease medications (yes/no) at randomization. Covariates, such as baseline score, were included in this model to ensure that the analysis results were evaluated based on the same adjusted baseline level. Efficacy comparisons for this model were based on the treatment difference versus placebo using least squares means and hypothesis testing utilized a one-sided α of 0.05, corresponding to a statistical significance threshold of p < 0.1 in two-sided values. PGI-I and MCGI-I scores were analyzed using a Cochran–Mantel–Haenszel test. C-SSRS, QUIP-RS, and PWC-20 findings were summarized descriptively.
Results
Subjects
This study was terminated early. However, the decision to terminate was not based on, and did not consider, data from the study itself. Prior to the start of this study, a strategic linkage was established with a concurrent study in more advanced PD (ClinicalTrials.gov NCT02687542). When the interim analysis of data from the more advanced PD study showed a low probability that it would reach a target efficacy threshold, (which was not a comparison with placebo, but a more stringent placebo-adjusted target value) that study was terminated, triggering a decision to terminate this trial early as well. Screening and randomization into this study were stopped, but subjects already randomized at the time of termination were permitted to complete all visits. Overall, 57 subjects were randomized and received study treatment (placebo = 28 and PF-06649751 = 29; Figure 2). Of these, 22 (78.6%) and 25 (86.2%) completed the study in the placebo and PF-06649751 groups, respectively.
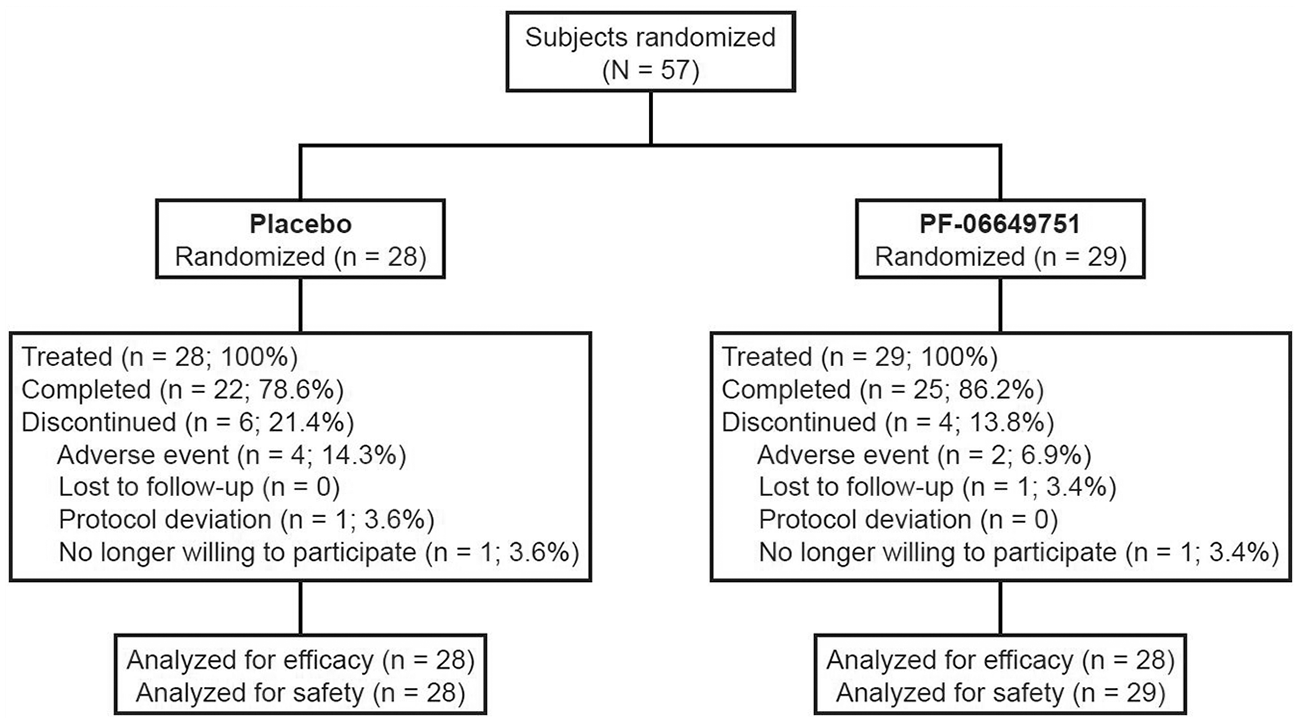
Subject disposition.
Most subject demographics were similar between treatment groups, although a higher male to female ratio was evident in the PF-06649751 group relative to placebo (Table 1). The mean duration in years since Parkinson’s disease onset was similar in both groups (placebo = 1.61; PF-06649751 = 1.42). Concomitant PD medication use during the study was also comparable in the two treatment groups. Overall, 16/29 subjects in the PF-06649751 group and 16/28 subjects in the placebo group received concomitant medication to treat PD symptoms. The medications most frequently taken were rasagiline/rasagiline mesylate (taken by 13/29 subjects in the PF-06649751 group and by 10/28 subjects in the placebo group) and amantadine (taken by 4/29 subjects in the PF-06649751 group and 6/28 subjects in the placebo group).
Subject demographics and baseline characteristics.

BMI, body mass index; MDS-UPDRS, Movement Disorder Society Unified Parkinson’s Disease Rating Scale; SD, standard deviation.
Mean (SD) treatment compliance was high in both groups [placebo = 99.70% (1.00%); PF-06649751 = 98.25% (3.33%)]. Mean (SD) treatment duration was 87.9 (34.4) and 96.8 (23.2) days in the placebo and PF-06649751 groups, respectively. The number of subjects reaching the dose maintenance phase was 22 (78.6%) in the placebo group and 26 (89.7%) in the PF-06649751 group. The maintenance dose (defined as the dose received for at least the last 4 weeks of the maintenance phase) for subjects in the PF-06649751 group was as follows: 0.75 mg (n = 1), 1.5 mg (n = 1), 3 mg (n = 5), 5 mg (n = 1), 7 mg (n = 2), 11 mg (n = 4), and 15 mg (n = 11). One subject in the PF-06649751 group reached the dose maintenance period but never achieved a maintenance dose due to discontinuation of treatment. Otherwise, there were no dose adjustments during the maintenance period in either treatment group.
Efficacy
The PF-06649751 group exhibited a mean (SE) change, from baseline to week 15, in MDS-UPDRS Part III score of −9.0 (1.54) compared with −4.3 (1.65) for placebo (Figure 3). This corresponds to a least squares mean (SE) (90% CI) improvement over placebo of 4.8 (2.26) (1.0, 8.6) for the PF-06649751 group (2-sided p = 0.0407). PF-06649751, therefore, met the first predefined efficacy criteria by demonstrating an observed mean treatment effect over placebo of greater than 3.6 points. The probability that this observed treatment effect over placebo was greater than 0 was determined to be 98%; thus, PF-06649751 also met the second prespecified primary efficacy criteria and the overall primary endpoint. Statistically significant improvements in MDS-UPDRS Part III scores were also evident in the PF-06649751 group, compared with placebo, at all timepoints prior to week 15 (weeks 3, 5, 7, 9, and 12; all 2-sided p < 0.1; Figure 3).

Change in MDS-UPDRS Part III score over the course of the study. Data shown are mean ± SE. 2-sided p ⩽ 0.1 versus placebo; 2-sided p value at week 15 (primary endpoint) was 0.0407.
Exploratory analyses demonstrated statistically significant (2-sided p < 0.1) improvements in combined MDS-UPDRS scores (Part II + III, Part I + II + III, and total) at all timepoints assessed (weeks 9 and 15) in the PF-06649751 group compared with placebo (data not shown), driven in large part by changes in MDS-UPDRS Part III score. Exploratory assessment of PGI-I scores at weeks 9 and 15 demonstrated statistically significant improvement (lower score) in the PF-06649751 group compared with placebo. Mean/median PGI-I scores were 3.0/3.0 in the PF-06649751 group at week 9 compared with 3.5/4 for placebo (2-sided p = 0.055) and 2.5/2.0 in the PF-06649751 group at week 15 compared with 3.2/3.5 for placebo (2-sided p = 0.039). However, there was no statistically significant difference between treatment groups for any of the following scales at weeks 9 and 15: MDS-UPDRS Part I, MDS-UPDRS Part II, PDQ-39 global score, EQ-5D-5L, OSDMT, MCGI-I, MCGI-S, and PGI-S (all data not shown).
Safety
There was no apparent difference between treatment groups on the BDI-II, ESS, QUIP-RS, C-SSRS, and PWC-20 at weeks 9 and 15 (data not shown).
Treatment-emergent AEs were reported in 86.2% (n = 25) and 64.3% (n = 18) of subjects in the PF-06649751 and placebo groups, respectively (Table 2). Rates of withdrawal due to AEs were 6.9% for the PF-06649751 group and 14.3% for placebo. One serious AE (suicidal ideation) occurred in a subject in the PF-06649751 group. This event started and resolved on day 64, but led to study discontinuation on day 70. Per the Columbia Suicide Severity Rating Scale that was performed on Day 71, this subject had suicidal ideation (nonspecific active suicidal thoughts, but active suicidal ideation with any methods and no intent to act). In addition, this subject reported depression beginning on day 64 and resolving on day 81. No deaths were reported. The most common all-causality AEs in the PF-06649751 group were nausea (31.0%), headache (24.1%), dry mouth (17.2%), somnolence (13.8%), and tremor (13.8%).
Summary of AEs.

AEs, adverse events.
All causality AEs occurring in >5% of subjects in either group.
More subjects treated with PF-06649751 met categorization criteria for standing diastolic BP decrease of ⩾20 mmHg (8 versus 2), standing systolic BP decrease ⩾30 mmHg (4 versus 1), supine diastolic BP decrease ⩾20 mmHg (9 versus 1), and supine systolic BP decrease ⩾30 mmHg (5 versus 0) than subjects treated with placebo. Regarding orthostatic hypotension, there was a difference between treatment groups for the incidence of diastolic BP postural difference >10 mmHg (3 for PF-06649751 versus 1 for placebo) and there was no apparent increase in orthostatic hypotension-related AEs such as dizziness, fainting, or lightheadedness. No subjects met criteria for significant ECG changes.
There were no clinically meaningful effects of PF-06649751, relative to placebo, for standard laboratory safety assessments, including blood chemistry and liver function.
Discussion
Prior clinical studies of PF-06649751 in Parkinson’s disease were supportive of continued study based on results of MDS-UPDRS assessment, but were limited in both duration and sample size. The present 15-week study adds to the emerging efficacy and safety dataset of PF-06649751 in terms of number of subjects and duration of dosing, but also by examining safety and efficacy in a new subpopulation of subjects with Parkinson’s disease. Specifically, this is the first study of PF-06649751 in subjects with early stage Parkinson’s disease, as previous phase I studies enrolled subjects with more advanced Parkinson’s disease on a stable dose of at least 300 mg/day levodopa. In addition, this phase II study incorporated a new flexible dosing paradigm and had a longer stable dosing period than other PF-06649751 studies conducted to date.
The target MDS-UPDRS Part III efficacy threshold for PF-06649751 (change from baseline relative to placebo) in the current trial was 3.6 points, and was based on an internal meta-analysis of historical studies in subjects with early Parkinson’s disease, which utilized the traditional Unified Parkinson’s Disease Rating Scale (UPDRS). The minimal clinically important difference in traditional UPDRS Part III score is considered to be approximately 2.5 points. 33 Therefore, we sought to set our efficacy criteria as PF-06649751 having at least a 3 point improvement over placebo in traditional UPDRS Part III score after 15 weeks of treatment. This corresponds to an improvement of at least 3.6 points in the newer MDS-UPDRS Part III. 34 PF-06649751 met the primary study endpoint in this trial by improving MDS-UPDRS Part III score, from baseline to week 15, by 4.8 points on average more than placebo, exceeding the prespecified criterion of 3.6 points. Statistical improvement over placebo was evident from the first assessment (3 weeks), suggesting a rapid onset of action of PF-06649751 that requires further study for confirmation.
The improvement in motor function observed in this phase II study is in broad agreement with assessments of PF-06649751 efficacy in two previous phase I/Ib trials in subjects with Parkinson’s disease. In one of these trials, a single 9-mg dose of PF-06649751 significantly improved MDS-UPDRS Part III score, compared with placebo, at 1, 2, 4, 8, and 12 h postadministration. Improvements were also evident for 3 mg PF-06649751, though differences from placebo were not statistically significant. 24 In the other trial, sustained reductions from baseline in MDS-UPDRS Part III score were evident in subjects receiving once-daily, open-label, administration of increasing doses of PF-06649751 (at target doses of 5, 15, and 25 mg). 24
Though PF-06649751 met the primary efficacy endpoint, some limitations of the study should be noted. Foremost, enrollment in the study was terminated early by the sponsor. Overall, 57 subjects were enrolled instead of the 88 that were planned. This was not due to safety concerns, but rather to the results of an interim analysis conducted on a separate, phase II trial of PF-06649751 up to 15 mg in subjects with advanced Parkinson’s disease (ClinicalTrials.gov: NCT02687542). The primary analysis in that phase II trial was based on a specific, prespecified, target level of efficacy for PF-06649751 over placebo, which was deemed unlikely to be achieved by the interim analysis. However, as that study was terminated at approximately 50% enrollment, there was insufficient data to make any firm statistical conclusion about PF-06649751 efficacy versus placebo. In addition to the target levels of the primary analysis, there are several other factors that may explain the apparent discrepancy in efficacy between these studies, including the stage of Parkinson’s disease (early versus advanced), dosing approach (flex-dosing monotherapy versus fixed-dose add-on), length of titration (9 weeks versus 3 weeks), primary endpoint (MDS-UPDRS Part III versus subject reported OFF time), and other operational aspects. It should be noted that, although PF-06649751 consistently and significantly improved MDS-UPDRS Part III scores during the current trial, the trial was focused on subjects with early stage Parkinson’s disease and there remains a need to assess the safety and efficacy of PF-06649751 in other stages of the disease.
The overall safety profile of flexible-dose PF-06649751 (up to 15 mg) in subjects with early stage Parkinson’s disease was similar to prior studies of PF-06649751, with the majority of AEs being mild or moderate in severity. 24 The most commonly observed AEs in the PF-06649751 group were nausea, headache, dry mouth, somnolence, and tremor. In addition, consistent decreases in systolic and diastolic blood pressure, increases in pulse rate, and modest, but not clinically significant, changes in ECG parameters were evident in the PF-06649751 group relative to placebo.
The findings from this study suggest that PF-06649751 provides improvement of motor function, and is generally well tolerated, in subjects with early stage Parkinson’s disease. This warrants further study of PF-06649751 in subjects with Parkinson’s disease, and provides additional rationale and support for the continued development of this D1-selective partial agonist.
Footnotes
Acknowledgements
We thank Frederick Carter (Syneos Health) for critical review of the manuscript.
Author contributions
RR, YZ, SD, and DG conceived the research project, which was organised by JW, YZ, SD, and DG, and executed by RR and JW. Statistical analysis was designed by JW, YZ, SD, and DG, executed by YZ and SD and reviewed and critiqued by RR, JW, YZ, SD, and DG. A first draft of the manuscript was written by MS of Engage Scientific Solutions based on the clinical study report and discussion with all authors (see relevant conflicts of interest section). MS did not contribute to the design of the research, analysis of data, interpretation of data, or approval of the manuscript. The manuscript was reviewed and critiqued by RR, JW, YZ, SD, and DG, and approved by RR, JW, YZ, SD, and DG.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Pfizer.
Conflict of interest statement
JW and YZ are full-time employees of, and own stock or stock options, in Pfizer Inc. DG and SD were full-time employees of Pfizer Inc. at the time the study was conducted, and own stock in Pfizer Inc. DG is named on the patent for PF-06649751 (though all rights are assigned to Pfizer Inc). RR has no potential conflicts to disclose. Medical writing support provided by Matt Soulsby of Engage Scientific was funded by Pfizer.
RR reports no financial relationships with commercial interests.
JW, and YZ have received salary as full-time employees of Pfizer Inc. and own stock in Pfizer Inc.
SD and DG have received salary as full-time employees of Pfizer Inc., and subsequently Cerevel Therapeutics, and own stock in Pfizer Inc.
Data sharing
Upon request, and subject to certain criteria, conditions and exceptions (see ![]() for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US or EU, or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US or EU, or (2) in programs that have been terminated (i.e. development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.