
Abstract
The introduction of genetics revolutionized the field of neurodegenerative and neuromuscular diseases and has provided considerable insight into the underlying pathomechanisms. Nevertheless, effective treatment options have been limited. This changed recently when antisense oligonucleotides (ASOs) could be translated from in vitro and experimental animal studies into clinical practice. In 2016, two ASOs were approved by the United States US Food and Drug Administration (FDA) and demonstrated remarkable efficacy in Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA). ASOs are synthetic single-stranded strings of nucleic acids. They selectively bind to specific premessenger ribonucleic acid (pre-mRNA)/mRNA sequences and alter protein synthesis by several mechanisms of action. Thus, apart from gene replacement, ASOs may therefore provide the most direct therapeutic strategy for influencing gene expression. In this review, we shall discuss basic mechanisms of ASO action, the role of chemical modifications needed to improve the pharmacodynamic and pharmacokinetic properties of ASOs, and we shall then focus on several ASOs developed for the treatment of neurodegenerative and neuromuscular disorders, including SMA, DMD, myotonic dystrophies, Huntington’s disease, amyotrophic lateral sclerosis and Alzheimer’s disease.
Keywords
Introduction
Substantial progress in genetics has provided considerable insights into the pathomechanisms of several neurodegenerative and neuromuscular disorders. However, the translation of this knowledge into clinical practice has been challenging, and, until recently, efficient treatment strategies were scarce. This changed in September 2016, when the United States US Food and Drug Administration (FDA) approved the new antisense oligonucleotide (ASO) eteplirsen for the treatment of Duchenne muscular dystrophy (DMD). Shortly after, in December 2016, the FDA approval followed for the ASO nusinersen for the treatment of patients with spinal muscular atrophy (SMA).
ASOs are synthetic single-stranded strings of nucleic acids that consist of only a few bases (8–50 bases, ‘oligo’). They bind complimentarily (‘antisense’) through Watson–Crick base pairing to a defined part of a nucleotide sequence of the premessenger ribonucleic acid (pre-mRNA) and mature mRNA (‘sense’) and modulate its function. 1 Thus, the treatment with ASOs may represent the highest target specificity and provide an opportunity for addressing previously inaccessible drug targets. 2
The history of ASOs 3 is intimately connected with the first description of ‘nuclein’ in cell nuclei by Miescher in 1869, 4 the discovery of deoxyribonucleic acid (DNA) as hereditary material in 1944, 5 and the molecular structure of nucleic acids. 6 In the 30 years thereafter, intensive efforts in basic research were made, and several approaches comprising chemical modifications7–9 as well as an automated synthesis of oligonucleotides10,11 paved the way for a therapeutic use of nucleic acids. In 1978, Zamecnik 12 and Stephenson 13 demonstrated that the addition of a 13-mer oligodeoxyribonucleotide that binds complementarily to Rous sarcoma virus RNA can inhibit protein expression in cell cultures. The first ASO investigated within a phase I trial was designed to target p53 transcripts in patients with acute myelogenous leukaemia. 14 This was followed by the first approval of fomivirsen in 1998, an ASO for the treatment of cytomegalovirus retinitis in patients with immunodeficiency. 15 In the field of neuroscience, the first ASO was used in vivo in the brain in 1993 and targeted the neuropeptide Y1 (NY1) receptor mRNA. 16 By repeated injections of this ASO in the cerebral ventricle of rats, a specific inhibition of NY1 receptor expression was observed and was accompanied by behavioural alterations (e.g. anxiety). A few months later, another study reported that an ASO targeting the mRNA of N-methyl-D-aspartate receptor 1 (NMDA-R1) protein in rats selectively suppressed protein translation in vivo and prevented neurotoxic effects after cerebral ischaemia. 17 These results further supported the applicability of ASOs to neurological disorders.
Depending on their chemical design and target, ASOs exhibit their effects through a diverse set of mechanisms that have been extensively discussed in previous reviews.2,18,19 In general, with regard to their mechanism of action, ASOs can be categorized into those that promote RNA degradation and those that do not. RNA-degrading ASOs recruit endogenous enzymes such as ribonuclease H (RNase H), an enzyme that recognizes RNA–DNA heteroduplexes and cleaves the RNA strand. The binding of the ASO to its target mRNA mimics this DNA–RNA pairing. Thus, the cleavage of the target mRNA by RNase H leads to a reduction of the corresponding protein.18,20 Other common mechanisms of ASOs for reducing the amount of protein comprise translational inhibition or alterations of RNA stability via RNA modification. 18 There, ASOs pair with the target mRNA but, given their chemical design, they do not initiate mRNA degradation. For example, ASOs can bind to mRNA structures and prevent the 5’-mRNA cap formation or, alternatively, they modify the polyadenylation site to prevent mRNA translation or alter RNA stability. Moreover, ASOs can directly stick to the mRNA and sterically block the 40S and 60S ribosomal subunits from attaching or running along the mRNA transcript during translation. 19 Other ASOs bind on pre-mRNA intron/exon junctions and directly modulate splicing by masking splicing enhancers and repressor sequences, skipping exons, or forcing the inclusion of otherwise alternatively spliced exons.19,21–23 ASOs can also be designed to directly bind to microRNA (miRNA) sequences and inhibit the binding of their own target mRNA. 24 In addition, some ASOs bind to natural antisense transcripts (NATs). NATs are regulatory endogenous RNAs that are complementary to other endogenous RNA strands.25–27 By various regulatory mechanisms including the direct pairing with the sense transcript, they facilitate or reduce protein expression. 27 Thus, the administration of an ASO that antagonizes a NAT, for example, prohibits the NAT from inhibiting their mRNA and thereby, increases the corresponding protein levels. 28 A summary of these basic principles is depicted in Figure 1.
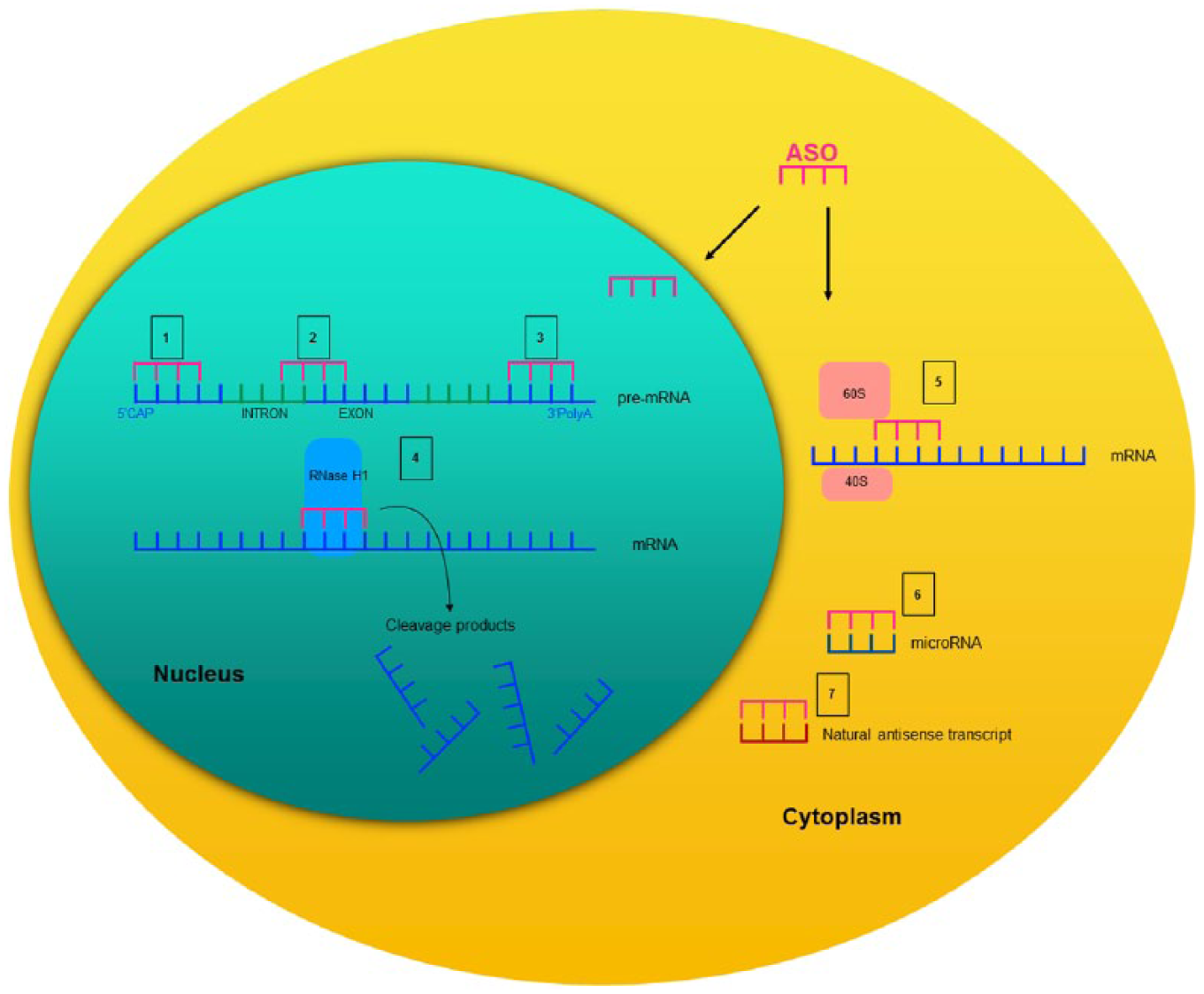
Schematic description of several mechanisms of action of synthetic antisense oligonucleotides.
The development of ASOs for clinical application was challenging because unmodified oligonucleotides are inherently unstable and are rapidly degraded by ubiquitously expressed endo- or exonucleases. 29 As such, several chemical oligonucleotide modifications were required that, in sum, increased their ability to recognize the target mRNA (target specificity), their resistance to nucleases, their plasma half-life and their distribution to tissues.2,30 The first and most remarkable chemical alteration, often referred to as ‘first generation of oligonucleotide modification’, was the modification in the phosphate backbone.2,18 One of the nonbridging oxygen atoms in the phosphate group was replaced by a sulfur atom, resulting in a phosphorothioate (PS) backbone.9,19,31 The addition of sulfur enhanced their nuclease resistance and elevated their plasma protein binding, most likely owing to the increased negative charge density of the sulfur backbone. 32 Apart from a better cellular uptake, 32 the phosphorothioate backbone modification supports the activation of the RNase H to break down target mRNAs, a critical mechanism of action of many ASOs. 2 Owing to their interaction with proteins, however, first-generation ASOs generate various nonspecific side effects, such as immune stimulation and complement activation. 33 Another backbone modification comprises the substitution of the 3’ oxygen in the deoxyribose ring with a 3’ amino atom. The thiophosphoroamidates and phosphoramidates exhibit high affinity toward complementary RNA and high nuclease resistance.2,34 However, they do not activate RNase H, a feature that makes them more suitable for nondegrading RNA manipulations, such as the modulation of splicing or translation inhibition.2,19 A second generation of ASOs were modified on the 2’ position of the sugar moiety. The most commonly used are 2’-O-methyl (2’-O-Me) and 2’-O-methoxyethyl (2’-MOE) oligonucleotides. They probably provided the highest value in the development of ASOs owing to their resistance to nucleases and their increased binding affinity, which is mostly driven by the electronegative substituent at the 2’ position. 33 Though, the most considerable disadvantage of the 2’ modification is their restricted ability to activate RNase H. 2 This motivated the development of chimeric ASOs, in which a core of a PS backbone is flanked by nuclease-resistant arms consisting of 2’-O-Me or 2’-MOE oligonucleotides. 33 The resulting ‘gapmer’ enables the cleavage of the target mRNA by RNase H at the central region of the ASO, whereas the outer portions provide an increased nuclease resistance and binding affinity.2,19,33 Other 2’ modifications comprise locked nucleic acids (LNAs), in which 4’-carbon has been tethered to the 2’-hydroxyl group. Although LNA oligonucleotides exhibit an increased nuclease resistance and binding affinity, they do not support RNase H and here, as well, a gapmer design has to be employed. 35 In addition, an increased hepatotoxicity has been reported for LNA ASOs. 36 This toxicological property can be decreased by combining the structural elements of 2’-MOE and LNA nucleosides and, thus, as reported, yielded a series of modifications. One of these comprised the 2’,4’-constrained ethyl (cEt) modification that possesses a binding affinity similar to LNA ASOs by an otherwise improved safety profile. 37 Other backbone modifications were developed by the implementation of a morpholine rather than a ribose ring in oligonucleotides. These ASOs, referred to as morpholino ASOs, 2 are resistant to nucleases and are less likely to interact with proteins owing to their neutral charge. 38 However, they do not activate RNase H and, therefore, are primarily used in translation arrest. 2 Various recently developed modifications and gapmer designs that have improved pharmacokinetic and pharmacodynamic properties of ASOs have been discussed in considerable detail in other reviews.1,2,18,19
Although these chemical modifications have improved drug-like properties and specific mechanisms of action, the implementation of ASOs in the treatment of multiple neurological disorders is still challenging. The drugs have to overcome membrane barriers that can be regarded as one limiting step. ASOs do not efficiently cross an intact blood–brain barrier (BBB). 2 Both their size and charge prevent their distribution across the BBB, and several studies have shown that fewer than 1% of systemically administered oligonucleotides reach the brain.30,39,40 Thus, efforts to achieve an efficient delivery to the central nervous system (CNS) are ongoing. One mechanism by which an ASO can cross the vascular barrier is via receptor-mediated endocytosis. 41 A biotinylated ASO captured with a streptavidin-conjugated monoclonal antibody to the mouse transferrin receptor reached the brain by this mechanism. 42 The same receptor-mediated endocytosis pathway was used to deliver nanoparticles carrying ASOs into the brain parenchyma. 43 Another mechanism was demonstrated by cell-penetrating peptide (CPP)-based delivery systems. 44 CPPs are 5–30 amino acids’ long, positively charged peptides that transport various macromolecules across cell membranes.45,46 In mice, systemically delivered ASOs tagged with arginine-rich CPPs were able to cross the BBB and were thereafter widely distributed in the brain. 47 Nonetheless, owing to their different chemical properties, not all ASOs are suitable for CPP coupling. ASOs encapsulated in exosomes are also able to cross the BBB, as shown by an intravenous injection of exosomes that were transduced with short viral peptides derived from rabies virus glycoprotein. 48 Recently, other viral vectors have been developed to deliver ASOs. To date, the adeno-associated virus vector possesses the most favorable properties because of it seems to be nonpathogenic and exhibits a high carrying capacity. 49 Apart from promising research to facilitate BBB penetration after systemic, intranasal, or oral administration, most of the currently available ASOs for the treatment of neurological disorders must be applied by intracerebroventricular or intrathecal delivery. Nevertheless, ASOs may provide the most direct treatment strategy for several neurological disorders. Given their well-characterized underlying pathogenetic mechanism and mutations within single or only a few genes, the potential efficacy of ASOs in the following disorders is plausible.
Spinal muscular atrophy
Spinal muscular atrophy (SMA) is a genetic autosomal-recessive disease with a progressive degeneration of alpha motor neurons. It results in proximal spinal and bulbar muscle weakness and atrophy. Consecutive impairments of respiratory muscles often lead to respiratory failure and reduced life expectancy. With an incidence of 1:10,000, SMA is the most frequent autosomal-recessive lethal disease in children after cystic fibrosis. 50 The clinical phenotype of SMA can be classified into four different subtypes (SMA type I–IV) ranging from severe to mild depending on the age of onset and level of motor function. 51 The most severe SMA type I (Werdnig–Hoffmann disease) accounts for about 60% of all cases. 50
SMA is most frequently caused by a homozygous deletion or mutation within the survival motor neuron 1 (SMN1) gene located on chromosome 5q13. Apart from these ‘5q-associated SMA types’, several other gene mutations have been observed that lead to clinically heterogeneous types of SMA. 52 The SMN1 gene codes for the ubiquitously expressed ‘survival motor neuron’ (SMN) protein, particularly important for the functioning of alpha motor neurons.51,53,54 Due to evolutionary gene doubling, 55 humans have one or more paralogous SMN1 gene copies, referred to as SMN2, which differ from SMN1 only by a few bases. Specifically, a cytosine-to-thymine mutation in exon 7 of the SMN2 gene leads to alternative splicing processes with the consequence that exon 7 is omitted from the majority of SMN2 transcripts. This results in a small amount (approximately 10%) of functional SMN protein expressed via the SMN2 gene. 51 Though, the clinical phenotype of SMA has been proven to be influenced by the number of SMN2 copies 56 and symptom severity inversely correlates with the number of SMN2 gene copies. Specifically, the presence of more copies of SMN2 leads to an increased production of normal SMN protein and a less severe clinical phenotype.56,57
Previous treatment options in SMA had been restricted to the clinical management of pulmonary, gastrointestinal, nutritional, and orthopaedic complications. 58 This changed with the introduction of the ASO nusinersen and its approval by the FDA in December 2016 for all 5q-associated SMA types. The approval by the European Medicines Agency followed in June 2017. The fully 2’-MOE PS antisense drug nusinersen 59 specifically binds to a sequence in intron 7 of the SMN2 pre-mRNA. The drug hereby enhances the inclusion of exon 7 in the mRNA by the inhibition of negative splicing factors, resulting in an increased amount of SMN protein.60,61
Within an open-label phase I study, 62 28 patients with SMA type II or III were investigated under four ascending single-dose levels of nusinersen (6 patients each with 1, 3 and 6 mg; 10 patients with 9 mg) to test drug safety and tolerability, pharmacokinetic properties, and preliminary clinical efficacy [ClinicalTrials.gov identifiers: NCT01494701 and NCT01780246]. Beyond the fact that the drug was well tolerated, this study provided early evidence for the clinical efficacy in humans by demonstrating a dose-dependent increase in motor functioning after a single intrathecal dose. Another phase II study 59 investigated the effects of 6 or 12 mg nusinersen in 20 infants with SMA type I over a period of 2–32 months. A significant improvement in the 12 mg-dose group was observed regarding the development of motor milestones and functions, survival, and the use of permanent assisted ventilation compared with baseline or with the natural history of the disorder [ClinicalTrials.gov identifier: NCT01839656]. Of the 20 patients, 4 died during the study, but this seemed to be associated with the natural history of the disease rather than with the side-effect profile of nusinersen. Investigations of autopsied neuronal tissues in three of these four patients revealed a two- to sixfold increase in SMN2 transcripts, including exon 7, as well as an increase in SMN protein compared with that known in untreated infants with SMA. Thus, the results provided evidence for an efficient correction of the SMN2 splicing by nusinersen in humans that previously had been observed only in animals.
A recently published phase III study 63 [ClinicalTrials.gov identifier: NCT02193074] investigated 121 symptomatic patients with SMA type I in a randomized, double-blind, and sham-procedure controlled design, in which two thirds of the infants received 12 mg nusinersen on days 1, 15, 29 and 64, and every 4 following months afterwards. Interim analyses showed that 41% of individuals treated with nusinersen achieved clinically significant improvements in motor functions after 13 months compared with sham-procedure controls (0%). This result prompted early termination of the trial and a submission to the FDA. In the final analyses, an even higher percentage of infants (51%) achieved motor milestones in the nusinersen group compared with controls. These results were confirmed by another randomized, double-blind, sham-procedure controlled phase III study 64 [ClinicalTrials.gov identifier: NCT02292537] in 126 symptomatic patients with SMA type II and III. Significant improvements in motor functions were demonstrated in children treated with nusinersen compared with the control group, in which motor functions decreased over 15 months corresponding to the natural history of the disease. Owing to the positive interim analysis, this study was also halted. An ongoing study is evaluating the long-term safety and tolerability of nusinersen in patients with SMA who previously participated in investigational studies of this ASO [ClinicalTrials.gov identifier: NCT02594124]. An additional phase II study 65 [ClinicalTrials.gov identifier: NCT02386553] is currently investigating the effects of nusinersen in SMA patients, in whom clinical symptoms were not evident at the beginning of the study. The observation in animal studies that the earlier the SMN levels were augmented, the better the therapeutic outcome, inspired this study.61,66 Interim analysis points to promising results: the 20 presymptomatic infants with SMA have reached motor milestones after a median of 317 days treatment. These results represent a breakthrough in the treatment of SMA type I in view of the fact that such achievements in motor functions are nearly comparable with those observed in healthy infants.
The approval of the ASO nusinersen not only represents a successful approach to the treatment of SMA, it also represents a proof of concept to successfully translate antisense technology from the bench to the clinic. Nevertheless, several caveats have to be taken into account. First, nusinersen cannot efficiently cross the BBB and has to be administered into the intrathecal space by lumbar puncture. Second, nusinersen has to be given in a standard dose of 12 mg on days 0, 14, 28, and 63 followed by repeated applications in 4-month intervals. To date, no specific side effects for nusinersen are known; but renal or hepatic dysfunctions, as well as undesirable effects on coagulation and complement or antibody activation, have to be considered, as is the case for other ASOs, especially after systemic administration.2,30,67–69
In addition, the encouraging results of nusinersen treatment of SMA are based on clinical studies in infants. The clinical efficacy of nusinersen in adolescents or adults with less severe disease progression (e.g. SMA type III or IV) has not yet been studied. The identification of sensitive parameters to monitor drug efficacy in such individuals will pose a considerable challenge.
Despite these caveats, various genetic alterations contribute to the broad spectrum of clinical phenotypes in SMA 52 and make concomitant targeting of several pathological mechanisms in SMA plausible. 70 However, even a combination of drugs directed at just the SMN pathway have also proven more effective than an individual treatment, as shown by an ASO that targets the SMN2-repressing long noncoding RNA (lnc-RNA). 71 The lnc-RNA arises from the antisense strand of SMN (SMN-AS1) and represses SMN expression. A degradation of SMN-AS1 with ASOs increased SMN expression in patient-derived cells, cultured neurons, and the CNS in mice. Notably, when the SMN- AS1 targeting ASO was delivered in combination with SMN2 splice-switching ASOs, the SMN expression was additively increased and survival improved.
Another novel approach to the therapy of SMA goes even a step further and entails gene replacement rather than the pure alteration of gene expression. Mendell and colleagues 72 recently reported about their open-label phase I/II study [ClinicalTrials.gov identifier: NCT02122952], in which 15 patients with SMA type I received a single dose of an intravenous nonreplicating adenovirus vector that included the normal human SMN1. The treatment was well tolerated and resulted in longer survival, superior achievement of motor milestones, and better motor functions than observed in historical cohorts. This also may reflect the start of an era of novel therapies in the field of neurology.
Duchenne muscular dystrophy
DMD is a genetic X-linked recessive muscular disease clinically characterized by progressive muscular wasting and weakness within the first years of life, and loss of the ability to walk during childhood or adolescence. Owing to respiratory failure or cardiac dysfunction, the disorder leads to death in late adolescence,73,74 if patients are not supported by intervention strategies, such as non-invasive ventilation. DMD occurs with an incidence of 1:3500 male births and represents the most common inherited paediatric muscle disorder.75,76 The disease is caused by mutations in the dystrophin gene located on Xp21, resulting in truncated protein or loss of transcript through nonsense-mediated decay.2,77 Approximately 60% of dystrophin mutations are large insertions or deletions that shift the reading frame, whereas about 40% of cases are caused by point mutations.78,79 The majority of patients with DMD lack the dystrophin protein which plays a critical structural role in skeletal and cardiac muscles. The protein links the internal cytoskeleton to the extracellular matrix as a major component of the dystrophin glycoprotein complex (DGC).76,80 A clinically milder form has been referred to as Becker muscular dystrophy (BMD) and is caused by a reduction of the amount or alterations in the size of the dystrophin protein. 79
Previous treatment options for DMD have included nonpharmacological strategies, such as non-invasive ventilation, physiotherapy, and orthopaedic treatment. With regard to pharmacological treatment, only glucocorticoids have proved to be efficient in slowing down disease progression. 81 Major efforts has been devoted to developing efficient treatment strategies in DMD, ranging from gene replacement, stem-cell therapy, and aminoglycoside antibiotics and proteasome inhibitors. 79 Similar efforts have been made to develop ASOs that promote the skipping of specific dystrophin exons.
However, given that multiple genomic alterations cause DMD, no single ASO will appropriately address all forms of the disease. 2 Therefore, we focus here on the two most advanced ASOs in clinical trials, drisapersen and eteplirsen, both of which promote the skipping of exon 51. In general, skipping this exon restores the reading frame of dystrophin RNA and enables the translation of a truncated dystrophin protein as opposed to no dystrophin.2,82,83 This, in turn, has the potential to convert the severe DMD into a milder BMD phenotype.84,85 Notably, skipping exon 51 could help most patients, that is, approximately 14% of those with DMD. 86
The ASO drisapersen is a 2’-O-Me PS oligonucleotide that selectively binds to a sequence within exon 51 to promote exon skipping.2,83,87 An exploratory, double-blind, placebo-controlled phase II study 87 [ClinicalTrials.gov identifier: NCT01153932] has examined the safety and efficacy of drisapersen (6 mg/kg) in DMD. The ASO was administered either continuously (once weekly) or intermittently (nine doses over 10 weeks) by subcutaneous injections. After 25 weeks, significant improvement was observed for the primary endpoint (6-minute walk distance, 6MWD) when compared with baseline and with placebo in the continuously treated group. In contrast, no differences in 6MWD from baseline were observed under intermittent drisapersen administration compared with the placebo group. The most common adverse events reported were injection-site reactions and renal events, for example, subclinical proteinuria. However, the encouraging clinical effects under the continuous treatment with drisapersen compared with placebo did not reach statistical significance by week 49.86,87 A subsequent and larger double-blind phase III study investigated 186 patients randomized to drisapersen (6 mg/kg/week) or to placebo for 48 weeks. This study confirmed the lack of significant effects on primary endpoints (6MWD) with the consequence that drisapersen failed to receive market approval.2,86
Eteplirsen is a morpholino ASO that binds similarly to drisapersen on exon 51 of the dystrophin pre-mRNA. 88 An initial open-label study investigated six dosages of intramuscularly administered eteplirsen for safety and biochemical efficacy [ClinicalTrials.gov identifier: NCT00159250]. Elevated dystrophin expression was observed in muscle biopsies for the highest dosages (0.9 mg) along with good tolerability.89,90 A double-blind placebo-controlled study was conducted to investigate eteplirsen’s ability to improve the distance walked in the 6MWD test. 75 Twelve DMD patients were randomized to receive 30 or 50 mg/kg/week eteplirsen by intravenous infusion or placebo. After 24 weeks of treatment, no significant improvement in the 6MWD was observed under eteplirsen compared with placebo. However, muscle biopsies revealed an increase of dystrophin-positive fibers up to 23% in the patients treated with 30 mg/kg eteplirsen, whereas no such increase was detectable in the placebo group. At week 25, patients who initially received placebo switched to an open-label treatment with either 30 or 50 mg/kg/week eteplirsen. After a further 24 weeks (48 weeks after the start of this study), subjects treated with 50 mg/kg eteplirsen revealed a stable or even increased performance in 6MWD compared with a decline in those patients initially treated with placebo. Notably, even greater increases in dystrophin-positive fibers were observed at this time point (52% and 43% in the 30 and 50 mg/kg cohort, respectively), thereby indicating that treatment duration rather than dosage may account for the beneficial effects of the drug. Finally, long-term follow-up investigations after 36 months confirmed the maintenance of ambulation in the majority of these patients. 91 Eteplirsen was approved by the FDA in September 2016 for the treatment of patients with DMD and with a confirmed gene mutation that is amenable to exon 51 skipping. However, as mentioned previously, the development of efficient ASOs in DMD is challenging because multiple genomic alterations may cause this disorder. As such, ongoing research regarding the therapy of DMD with ASOs is focusing on agents that support multiexon skipping. Thus, the applicability of these ASOs may increase to up to 50–70% of DMD cases. 92
Myotonic dystrophies
Myotonic dystrophies (DMs) are genetic autosomal-dominant inherited multisystem diseases with phenotypic core patterns comprising distal myotonia, proximal muscular dystrophy, cardiac conduction defects, posterior iridescent cataracts and endocrine disorders, such as male hypogonadism or diabetes. 93 Based on their clinical phenotypes and the underlying genetic alterations, DMs can be assigned to two main subtypes. Steinert and colleagues first described in 1909 the ‘classic’ DM (now referred to as DM type 1) characterized by the clinical features described above. DM type 1 is caused by an expansion of an unstable CTG trinucleotide repeat in the 3’ untranslated region (UTR) of the myotonic dystrophy protein kinase (DMPK) gene located on chromosome 19q13.3 that codes for a myosin kinase expressed in skeletal muscle.93–96 Much later, a milder multisystem disorder was described with predominantly proximal rather than distal muscular weakness and cataracts, but without the respective gene defect described above.93,97–100 The pathomechanism is ascribed to an unstable tetranucleotide repeat expansion (CCTG) in intron 1 of the nucleic acid-binding protein (CNBP) gene on chromosome 3q21.101,102 Thus, patients with the clinical pattern of predominantly proximal myotonic myopathy with a positive test for unstable CCTG-repeat expansion on chromosome 3q21 are classified as having DM type 2. 103
Although DM types 1 and 2 share some clinical phenotypes, various dissimilar symptoms distinguish them. Whereas life expectancy is reduced in DM type 1, it is within the normal range in DM type 2. 93 DM is the most common adult-onset muscular dystrophy, and a large-scale population study revealed that DM type 2 may even be the most common inherited muscle disease in the European population. 104 Taken together, the prevalence of DM types 1 and 2 is estimated at 12–20 per 100,000, with widely varying rates in different populations.104,105
Currently, there is no cure for DM and the management in DM type 2 is similar to that in DM type 1 except for a few specific treatments. The main therapeutic options are restricted to supportive strategies: physiotherapy, assistive devices, such as wheelchairs, eye and cardiorespiratory monitoring, and the treatment of endocrinological abnormalities. 93 Several pharmacological treatments have been tested including mexiletine, gabapentin, nonsteroidal anti-inflammatory drugs, low-dose steroids, and tricyclic antidepressants but none of these has been effective. 93
However, there is increasing evidence for an effective treatment option with ASOs in DM. The mutations in DM type 1 and 2 induce mutant RNAs containing CUG or CCUG expanded repeats that are retained in the nuclei and alter alternative splicing factors, such as muscleblind-like 1 proteins (MBNL). The resulting misregulated splicing of several pre-RNAs appears to underlie most of the symptoms in DM. Based on the idea that a reduction of (CUG)n RNA dosage might be beneficial to patients, Mulders and colleagues 106 investigated the 2’-O-Me PS ASO (CAG)7 that silences mutant DMPK RNA expression and selectively minimizes the number of ribonuclear aggregates. In a DM type 1 mouse model, they observed a significant reduction of the level of toxic (CUG)n RNA and normalizing effects on aberrant pre-mRNA splicing as a proof of principle in vivo. Another study performed in a transgenic mouse model of DM type 1 investigated the effects of several systemically administered ASOs and also demonstrated a rapid decrease of CUG-expanded RNA in skeletal muscles by a 2’-MOE ASO along with normalized splicing and reversed myotonia. Impressively, the effect was sustained for up to 1 year after treatment discontinuation. 107
A further study investigated the effects of a novel high-affinity class of an ASO containing cEt modification. 108 The systemic delivery of this ASO to wild-type mice decreased DMPK mRNA levels by up to 90% in skeletal muscle. In transgenic mice and Cynomolgus monkey, a similar impressive inhibition of DMPK was observed in cardiac muscle (up to 50%) and multiple skeletal (up to 70%) muscles. The ASO was well tolerated and here, too, a sustained inhibition of DMPK mRNA levels in muscles was demonstrated up to 13–16 weeks post treatment. Beyond these encouraging results arising from animal studies, Ionis Pharmaceuticals Inc. has recently completed a phase I/IIa placebo-controlled study to assess safety and tolerability of multiple doses of ISIS-DMPKRx in adults with DM type 1 [ClinicalTrials.gov identifier: NCT023412011]. To date, the chimeric ASO ISIS-DMPKRx that contains both, 2’-MOE and cEt modifications was well tolerated 109 ; efficacy results will be reported in the near future.
Huntington’s disease
Huntington’s disease (HD) is a rare monogenetic neurodegenerative disorder that is clinically characterized by hyperkinetic involuntary movements, dystonia, behavioural abnormalities and cognitive decline. Clinical symptoms typically manifest between 30–50 years of age and invariably progress over time. The prevalence in the White population is estimated at 5–10 per 100,000. 110 The autosomal dominantly inherited disease is caused by an abnormal expansion of the CAG repeat in exon 1 of the huntingtin (HTT) gene located on the short arm of chromosome 4. 111 It codes for the HTT protein that is involved in a broad range of cellular functions, including neuronal survival 112 and axon stability. 113 CAG repeats above 40 cause the disease with full penetrance, whereas CAG repeats between 37–39 show incomplete penetrance. CAG expansions ranging from 6 to 26 repeats are considered normal. The so-called intermediate alleles, ranging between 27 and 35 CAG repeats, are instable during meiotic transmission. 114
The mutant HTT protein harbours an expanded polyglutamine tract, which mediates toxic gain of function effects including intraneuronal aggregation or abnormal interactions with other proteins. 115 The medium spiny neurons of the striatum are particularly vulnerable to these toxic effects. An initial loss of the indirect pathway that leads to hyperkinetic movements is followed by a loss of MSNs of the direct pathway, thereby leading to hypokinesia. 116 The reasons for this preferential vulnerability remain speculative, although a different distribution of dopamine D2 receptor expression is thought to be involved. 117
Whereas progress in understanding the underlying genetics in HD has been substantial since its first clinical description in 1872, no disease-modifying treatment options are available, and current medical management is restricted to supportive care. This may change with the advent of HTT-lowering ASOs. In 2012, Kordasiewicz and colleagues investigated the in vivo effects in several mouse models of a 2’-MOE PS ASO with RNase H-mediated degradation properties that targets mutant HTT mRNA. Notably, they could demonstrate that a transient infusion of the ASO into the cerebrospinal fluid of symptomatic HD mice significantly delayed disease progression and decreased HTT levels that persisted for longer than the HTT knockdown.18,118 Transgenic human HTT mRNA could be decreased by 50–80% in most brain regions, and survival was extended even when the ASO was applied to a more severely progressing mouse model. The safety profile was excellent. However, it has to be taken into account that this ASO not only reduced the expression of mutant HTT but also that of wild-type HTT. Although the study detected no major adverse events, further research focused on increasing the specificity regarding mutant alleles. Hence, Østergaard and colleagues 119 introduced ASOs that enhance single nucleotide discrimination up to 100-fold for mutant HTT allele over normal in patient cells and in a humanized mouse model of HD.
Despite the ongoing general debate about how well mouse models recapitulate human diseases, these studies provided proof of concept for pursuing ASO treatment in HD. Recently, a phase I/IIa clinical study to assess safety, tolerability, and the pharmacological properties of multiple ascending doses of an intrathecally applied ASO [ISIS 443139, IONIS-HTTRx; ClinicalTrials.gov identifier: NCT02519036] in patients with early-stage HD was completed. The formal publication of the results from this trial is expected during the first half of 2018, but according to an online press release, a dose-dependent reduction of HTT has been observed, along with a good safety and tolerability profile.
Amyotrophic lateral sclerosis
ALS is a severe late-onset neurodegenerative disease with a progressive dysfunction of the upper and lower motor neurons of the motor cortex and spinal cord. Clinical symptoms include fasciculations, spasticity, progressive muscle weakness, and muscle atrophy. The disease is characterized by a fatal course and, typically, death occurs within 2–4 years from the time of diagnosis, mainly due to respiratory failure.120–122 In Europe, the incidence of ALS is estimated with 1.7–3.0 cases per 100,000,123,124 although rates greatly differ in various parts of the world. 121
The precise pathomechanisms of ALS have not been fully clarified. Substantial evidence exists for a pivotal role of phosphorylated 43-kDA Transactivating Response Region (TAR) DNA-binding protein (pTDP-43) in ALS pathology.125,126 An aggregation and progressive accumulation of pTDP-43 in selected neurons and oligodendrocytes is thought to cause their loss of physiological function. 127 In line with the clinical spreading pattern, pTDP-43 pathology seems to disseminate by cell-to-cell transmission via axonal transport and through synaptic contacts from cortical neuronal projections to other brain regions and the spinal cord. 128
About 95% of ALS cases are believed to occur sporadically, whereas approximately 5–10% are inherited.124,129 Several gene mutations could be associated with both sporadic (sALS) and familial (fALS) ALS.130,131 The best studied gene associated with ALS is SOD1, located on chromosome 21q.131,132 More than 180 mutations in this gene have been reported (http://alsod.iop.kcl.ac.uk/Overview/gene.aspx?gene_id=SOD1) and account for up to 20% of fALS and up to 2–3% of sALS cases.133,134 SOD1 encodes copper/zinc superoxide dismutase (Cu/Zn SOD), a homodimeric metalloenzyme that binds copper and structural zinc ions and catalyses the dismutation of superoxide radicals. 135 Although the detailed mechanisms are not yet fully understood, mutant SOD1 forms are assumed to decrease conformational stability and promote misfolding of the protein. 136 For years, toxic gain of function mechanisms were considered to cause SOD-linked ALS rather than a loss of protein function. 132 More recently, however, there is evidence that SOD1 loss of function may also play a considerable modifying role. 137 Nevertheless, the development of ASOs targeting SOD1 appeared a logical step, considering that lowering the levels of mutant SOD1 protein is predicted to slow disease progression. 138 Accordingly, a 2’-MOE PS ASO that targets SOD1 mRNA significantly reduced SOD1 protein and mRNA levels throughout the brain and the spinal cord in rats after intrathecal administration. 139 When this ASO was given prior to symptom onset, as defined by retrospective analyses in an ALS mouse model, disease progression could be delayed and survival extended. 139 A randomized, placebo-controlled phase I trial [Clinicaltrials.gov identifier: NCT01041222] investigated this ASO at increasing doses in humans with SOD1-related ALS. It demonstrated an excellent safety and tolerability profile. 140 Moreover, a second-generation ASO compound BIIB067 (IONIS-SOD1Rx) is entering a phase I/II trial [ClinicalTrials.gov identifier: NCT02623699] to evaluate its safety, tolerability and pharmacokinetic properties in a single-dose cohort, followed by a multiple-dose cohort in SOD1-linked ALS.
More recently, expanded hexanucleotide repeats in a chromosome 9p21 open reading frame 72 (C9orf72) were identified as the most common genetic cause of ALS that account for approximately 30% of fALS and up to 5% of sALS cases.141,142 Specifically, a GGGGCC hexanucleotide expansion occurs between two five prime noncoding exons of C9orf72. The gene encodes an uncharacterized protein with a yet unknown function, 143 but it may have an important role in membrane trafficking and autophagy. 144 The underlying mechanism that mediates disease pathogenesis is not fully understood. A loss of normal C9orf72 function has been proposed and seems plausible, given the fact that impaired autophagy and endolysosomal degradation are implicated in neurodegenerative diseases. 145 In support of this hypothesis, an experimental knockdown of the zebrafish orthologue of C9orf72 was associated with axonopathy and motor deficits that could be rescued by expressing human C9orf72 mRNA. 146 In addition, a homozygous knockout of the worm orthologue of C9orf72 resulted in a degeneration of motor neurons along with motor deficits. 147 Despite the loss of function, two further distinct gain-of-function mechanisms have been discussed: first, the formation of repeat RNA aggregates (RNA foci) in neuronal nuclei and, second, the generation of toxic dipeptide-repeat (DPR) peptides.127,148,149 Research on the development of efficient treatment options is a high priority because the repeat expansion in C9orf72 is a major cause of both frontotemporal dementia (FTD) and ALS. 141
Donnelly and colleagues 150 investigated several ASOs that recruit RNase H or block the interaction between the repeat expansion and RNA binding proteins. They demonstrated a reduction of toxic RNA foci and restored normal gene expression markers. A suppression of several pathological features, including RNA foci and DPRs, was concordantly shown for ASOs in primary cortical neurons from mice. 151 These results were confirmed by a recent investigation of ASOs in experimental animals 152 that selectively targeted repeat-containing RNAs and also led to a rapid reduction in RNA foci and DPRs. Notably, the applied single doses of these ASOs were also accompanied by a sustained improvement of behavioural deficits. Further promising results were recently observed regarding ASOs targeting the ataxin-2 (ATNX2) gene, which is implicated in spinocerebellar ataxia type 2 and as a modifier gene in TDP-43-driven ALS. After a single administration of ASOs targeting ATNX2 into the CNS of transgenic mice, survival could be extended considerably. 153
Together with these encouraging results, other ASO strategies that have been tested in animal models (summarized in Tosolini and Sleigh 154 ) indicate that the development of ASOs in the therapy of ALS is not lagging far behind those recently approved by the FDA for SMA and DMD.
Alzheimer’s disease
Alzheimer’s disease (AD) is the most prevalent cause of dementia and represents one of the major healthcare challenges in the 21st century given that prevalence rates are thought to double every 20 years until at least 2050. 155 Clinical features range from progressive memory impairment and executive dysfunction to psychiatric symptoms that severely interfere with activities of daily life. Decades of research regarding the underlying pathomechanisms of AD have identified two major molecular markers, namely, the accumulation of the abnormally folded amyloid beta peptide (Aβ) and tau proteins in amyloid plaques and neurofibrillary tangles.156,157
Aβ-pathologies are associated with intracellular processing of the amyloid precursor protein (APP), a transmembrane protein with cleavage sites for alpha- (α-), β- and gamma- (γ-)secretases. The cleavage of APP directly by α- and then γ-secretases typically does not generate Aβ or lead to its reinternalization in endosomal compartments. 158 However, when APP is cleaved by β- and γ-secretases, the resulting Aβ-fragment oligomerizes to insoluble fibrils and plaques 18 that cause toxicity through several mechanisms, including microglial infiltration, the generation of reactive oxygen species and synaptic damage. 158 Several APP mutations have been identified in early-onset, autosomal dominant, fully penetrant AD. 158
An early study 159 investigated the effects of phosphorothioate-modified ASOs targeting the A region of the APP gene, on reversing elevated APP levels and behavioural impairments in mice. Three intracerebroventricular injections of the ASO reduced APP levels by 43–68% in specific brain regions along with improvement in learning and of memory deficits. Subsequently, an intravenously administered APP targeting ASO was investigated in mice and confirmed a significant reduction of AβPP-signal along with decreased neuroinflammation by otherwise improved learning and memory. 40 However, the poor ability of this ASO to cross the BBB and the restricted transferability attributable to the use of specific mouse models represent considerable limitations, and have delayed the advancement of ASOs targeting APP. 18
Genetic components have not only been associated with early but also with late-onset sporadic disease forms. In detail, the apolipoprotein isoform ApoE-ε4 confers a 2- to 12-fold risk for the development of AD in humans. 160 The development of ASOs that target the apolipoprotein E receptor 2 (ApoER2) seems intuitive, inasmuch as the receptor modulates APP localization and the processing that results in an increased Aβ production. 161 Physiological ApoER2 signalling requires amino acids that are encoded by an alternatively spliced exon 18 in humans (exon 19 in mice), and a deregulated splicing mechanism could be demonstrated in AD. Hence, Hinrich and colleagues 162 investigated an ASO that increases exon 19 splicing in mice and observed that a single dose corrected ApoER2 splicing for up to 6 months along with a potential improvement in synaptic function, memory, and learning.
A pivotal role in the pathogenesis of AD but also in other neurodegenerative disorders has been ascribed to tau proteins. The microtubule-associated protein tau is physiologically involved in the stabilization of microtubules, in intracellular signaling, and in neurogenesis. 163 The human brain typically expresses six isoforms of tau protein derived from a single tau (MAPT) gene. The isoforms differ based on alternative splicing within the N terminus and a repeat domain region of the tau MAPT gene. 18 An exclusion of exon 10 results in splicing products expressing tau with three microtubule-binding repeats (MTBRs), while the inclusion of an imperfect repeat region encoding exon 10 leads to the expression of tau containing four MTBRs. 164 Whereas normal human brain typically expresses approximately equal levels of tau protein with three and four MTBRs, an alteration of this ratio has been observed in several tauopathies. 165 In addition, other abnormal post-translational modifications, such as tau protein hyperphosphorylation, are crucial to the early pathogenesis and neuronal death in AD.157,164
When it is taken into consideration that the accumulation of hyperphosphorylated tau protein is directly associated with cognitive decline in AD, 166 a lowering of tau expression by ASOs could provide at least one therapeutic strategy. A recently conducted study investigated the effects of an ASO that selectively decreased human tau mRNA and protein in a mouse model of tauopathies. 167 Overall, the authors observed a decreased burden of phosphorylated tau, of hippocampal volume loss, and of neuronal death accompanied by an extended survival in experimental animals, including nonhuman primates. Another treatment strategy with ASOs in AD focuses on MAPT mutations that alter splicing processes of exon 10. A recent study demonstrated an efficient exon 10 skipping by a 2’-MOE phosphothioate ASO to bias towards a tau protein containing three MTBRs. In two mouse models expressing human tau, the ASO lowered tau containing four MTBRs without changing the total amount of tau protein.18,168
The advantage of the specific reduction of tau protein with four MTBRs lies in its selectivity and leaves the total amount of tau unaltered. Otherwise, decreasing the total amount of tau protein by ASOs represents a substantial therapeutic approach potentially applicable for other tauopathies, such as corticobasal degeneration, progressive supranuclear palsy, argyrophilic grain disease, frontotemporal dementia with parkinsonism, and Pick’s disease. Ionis Pharmaceutials Inc. is currently conducting a phase I/II study to assess the safety and tolerability as well as the pharmacological properties of an intrathecally administered 2’MOE ASO [ISIS 814907; IONIS MAPTRx; ClinicalTrials.gov identifier: NCT03186989] in patients with mild AD. This ASO targets MAPT mRNA to decrease the amount of tau protein irrespective of its isoform.
Costs, cost effectiveness and ethical implications
Parallel to the remarkable progress in the development of ASOs, there is an ongoing debate regarding the cost-effectiveness ratio and ethical implications. For eteplirsen, the costs were estimated at US$57,600 per month despite the doubts whether the small increases of dystrophin could affect clinical progression of DMD.169,170 Considering the price of US$125,000 per one injection of nusinersen, costs of the treatment of patients with SMA with this ASO amounts to US$750,000 for the first year and US$375,000 for every year afterwards. 171 The currently high prices will be impossible to bear by any healthcare system and some national insurers already declined 170 or restricted the covering of the costs, for example, to patients with infantile-onset SMA type 1. 171 Notably, data on long-term evidence of cost effectiveness are still missing and formal clinical but also economic impact analyses after 1–2 years of treatment may pose only one considerable approach. Moreover, the high costs raise the questions about patients that live in underinsured parts of the world and how they may have access to these drugs. Thus, global and cohesive strategies have still to be developed together with physicians, health insurers, pharmaceutical companies and policymakers.
Conclusion
At 25 years after ASOs were first used in vivo in the brain, several specific ASOs are now available or are being tested in clinical trials for the treatment of a variety of neurodegenerative disorders. In September 2016, the FDA approved the ASO eteplirsen for the treatment of DMD. Only a few months later, in December of the same year, FDA approval followed for the ASO nusinersen for SMA. Together, these developments emphasize the therapeutic potential of ASOs in the treatment of neurodegenerative disorders and prove that ASO strategies can be transferred from the bench into clinical practice. As summarized in this review, efficient ASOs are under development or have already been tested in clinical trials for the treatment of MD, HD, ALS, and AD. Beyond their encouraging results, new efficient ASOs may be anticipated in peripheral neurodegenerative disorders. A recent study 172 reported the effects of an ASO for the treatment of the inherited peripheral nerve disorder Charcot-Marie-Tooth disease and demonstrated an improved myelination, even when treatment was started after symptom onset.
In summary, the advent of ASOs represents a therapeutic milestone for several neurological disorders. This was barely conceivable a few years ago. Although ASOs exhibit excellent safety and tolerability profiles, further improvements are required. Most ASOs have to be delivered directly into the intrathecal space. Not surprisingly, there is an ongoing debate regarding the cost effectiveness of these drugs. 171 However, the SMA success story demonstrated that ASOs are effective and safe; they may even succeed in revolutionizing the entire field of therapeutic strategies in neurology.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: CDW received travel expenses for attending a meeting sponsored by Biogen and she cooperates with Hoffmann-La Roche. ACL received financial research support from AB Science, Biogen Idec, Cytokinetics, GSK, Orion Pharam, Novartis, TauRx Therapeutics Ltd. and TEVA Pharmaceuticals. He also has received honoraria as a consultant from Mitsubishi, Orion Pharma, Novartis, Teva and as an advisory board member of Biogen, Treeway, and Hoffmann-La Roche.