
Abstract
Ischemic stroke is a major cause of death. Besides the direct damage resulting from oxygen and glucose deprivation, sterile inflammation plays a pivotal role in increasing cellular death. Damaged-associated molecular patterns (DAMPs) are passively released from dying cells and activate the innate immune system. Thus, they take part in the direct and rapid activation of the inflammatory response after stroke onset. In this review the role of the most important DAMPs, high mobility group box 1, heat and cold shock proteins, purines, and peroxiredoxins, are addressed. Moreover, intracellular pathways activated by DAMPs in microglia are illuminated.
Introduction
Stroke is a major issue in terms of an increasing incidence and considering disability-adjusted life years (DALYs) lost. In 2030 up to 12 million deaths are expected. 1 Moreover, it is the leading cause of long-term disability. 2 Approximately 2 million neurons per minute die when exposed to ischemia. 3 Acute thromboembolic or local thrombotic occlusion of a cerebral artery accounts for 80% of strokes. Systemic thrombolysis and mechanic recanalization are so far the only acute treatments in these cases, but less than 20% of patients receive it due to restrictive inclusion criteria. Besides the direct brain injury resulting from ischemia and reperfusion, there is growing evidence that a rapid evolving sterile inflammation worsens infarct size in the first 1–3 days post stroke, and thus clinical outcome. 4 In immune-deficient mice, about 20–30% smaller infarct sizes were detected after experimental stroke induction. 5 Cells invade the ischemic brain in a specific temporal and spatial pattern. 6 In the first couple of days, neutrophils, macrophages and microglia predominantly infiltrate the ischemic tissue in large numbers, reaching their maximum around 3 days post ischemia. Even though smaller in numbers, other potentially highly immune competent lymphocyte subpopulations and dendritic cells accumulate in the ischemic tissue during the first days after the ischemic insult. Overall, the initial cellular inflammatory reaction follows a highly conserved pattern of sterile inflammatory reactions, which are also known from other organs. Whereas the detrimental effects of proinflammatory signals released by the innate immune system occur in the early course of stroke, 7 the role of the adaptive immune system is still controversial. 3 Interestingly, the immunologic response not only accounts for detrimental effects, but it also influences neuronal regeneration in the following weeks and months after brain infarction. 8 In the setting of infections by pathogens, the immune system protects the human body against danger in general, as introduced by Polly Matzinger in 1994. 9 These warning mechanisms are not only released by pathogens, they also encompass released or secreted molecules from injured cells, which are called damaged-associated molecular patterns (DAMPs). 10 In general, DAMPs can be divided into PAMPs (pathogen-associated molecular patterns) and alarmins, which categorize endogenous molecules. 11 Both protein [e.g. high mobility group box 1 (HMGB1), heat shock proteins (Hsps)] and nonprotein alarmins (e.g. adenosine triphosphate (ATP)) exist. Overall, DAMPs are promising candidates in stroke pathophysiology as several experimental data indicate their importance for the activation of the immune system early following ischemia. 2 In stroke, the sterile inflammation starts with the release of DAMPs from dying neuronal and non-neuronal brain cells, which then activate local immune cells (e.g. microglia) and perivascular endothelia cells via pattern recognition receptors (PRRs) including toll-like receptors (TLRs). The local immune reaction is amplified, which results in the massive invasion of systemic immune cells. 3 Besides the local immune response, activation of the immune system also has systemic consequences, since the phenomenon of a systemic immune suppression can be observed in stroke patients. This is associated with an increased susceptibility to bacterial infections such as pneumonia or lower urinary infections. 12 As depicted, the inflammatory response is a major potential target of future therapeutic treatments in stroke. In this review, we focus on the acute involvement of DAMPs as one of the first reactions after ischemia.
High mobility group box 1
The HMGB1 protein is a widely expressed nonhistone cytokine-like factor, which mainly stabilizes the chromosomes in the nucleus. 13 Under physiological conditions, it has a proangiogenic effect on endothelial cells and neurite outgrowth is stimulated by HMGB1 during embryogenesis. 14 HMGB1 is both passively released from necrotic cells and actively secreted by the innate immune system in case of infections. Interestingly, it does not diffuse in the extracellular matrix when cells undergo apoptosis because here HMGB1 binds irreversibly to the modified chromatin.15,16 HMGB1 appears early in sterile inflammation and initiates the production of proinflammatory mediators.14,17 Multiple extracellular receptors are activated by HMGB1, including the receptor for advanced glycation end products (RAGE) or TLR2 and TLR4, which are ubiquitously expressed by resident central nervous system (CNS) cells. 18 Additionally, HMGB1 possesses divergent functions in different redox states. 19 In positions 23, 45 and 106, three cysteine residues are located. A disulfide bond between C23 and C45 as well as the thiol form of C106 is obligatory for its tumor necrosis factor (TNF)-stimulating effects via TLR4.20,21 Overall, the reduced form is associated with chemotactic functions whereas the disulfide form is thought to be proinflammatory through the induction of nucleotide-binding domain, leucine-rich repeat, pyrin domain containing protein 3 (NLRP3) and nuclear factor κBIα (NFκBIα). 22
HMGB1 also plays a pivotal role in ischemic stroke. High levels of systemic HGMB1 were measured in serums of patients with cerebral ischemia. 23 Elevated HMGB1 levels correlate with severe stroke sizes.24–26 In the CNS, HMGB1 is upregulated after stroke and maintains the inflammatory process. 27 It is secreted by activated astrocytes and microglia, and stimulates the release of interleukin (IL)-1, TNFα, IL-6 and IL-8, as well as inducible nitric oxide synthase (iNOS) expression.28–31 An increased vascular and blood–brain barrier permeability is observed. 31 Moreover, matrix metalloproteinase 9 (MMP-9) is upregulated by HMGB1 via TLR4 and its induced cytokines as TNFα or IL-1β mediating cellular death after ischemic stroke. 31 A maturation process of the passively released protein in the blood circulation to a cytokine-stimulating isoform has been described. 32 Furthermore, HGMB1-induced activation of RAGE results in systemic lymphocytopenia after brain ischemia. 32 Locally, the amplification of the post-ischemic inflammation is also maintained by HMGB1.33,34 Leucocyte infiltration is stimulated as well as activation of microglia and amplification of metalloproteinases. The late treatment with amlexanox or an anti-HMGB1 antibody after ischemia appears to protect the brain from stroke-induced brain damage by the inhibition of the release of HMGB1. 35 Several studies demonstrated protective effects by pharmacological inhibition of HMGB1 or its pathway underlying the detrimental function of HMGB1 in the early stages following stroke.3,36–38 Reduced inflammation and stroke size are seen after inhibition of RAGE or TLR4.34,39 Contrary to that, HMGB1 also stimulates angiogenesis, neurite outgrowth and neuronal survival in the late period after stroke, and is actively secreted by astrocytes with positive signaling on endothelial progenitor cells. 31 In summary, HMGB1 has mainly detrimental effects in the early post-ischemic period, though HMGB1 activation also occurs to take part in stroke recovery mechanisms.
Heat shock proteins and cold shock domain proteins
Hsps belong to the group of molecular chaperones, which help guide the correct folding of proteins during their synthesis. They are produced when cells are exposed to elevated temperatures or any kind of injury as part of a feedback system, which detects misfolded proteins. 40 The two major chaperones are Hsp60 and Hsp70, which are located in the cytosol. Hsp70 acts before the protein leaves the ribosome whereas Hsp60 interferes after the protein has been fully synthesized. 40 The Hsp70 is the best studied Hsp in ischemic stroke. Depending on its location, opposite effects of Hsp70 have been described. Intracellular Hsp70 can decrease the signaling of proinflammatory factors such as the transcription factor NFκB, MMPs and reactive oxygen species (ROS). Located in the extracellular matrix, Hsp70 binds to TLRs on macrophages, dendritic cells and microglia, and thereby activates the proinflammatory NFκB and its associated pathways. It can also elicit the immune response of CD4+ und CD8+ T cells.41,42 Following brain infarction, Hsp70 can be detected in neurons, microglia, astrocytes and endothelial cells. TNFα and IL-1β were downregulated after Hsp70 overexpression in experimental stroke. 41 Increased intracellular Hsp70 protein expression appears to be cerebro protective in stroke studies of viral overexpression in neurons and glia, whereas Hsp70 knockouts worsen outcome. 42 Additionally, exogenously delivered Hsp70 has a neuroprotective function. This is demonstrated by intravenous injection of Hsp70 in rat, reducing the cerebral infarction zone.43–45 Dynamin is a downregulated protein by Hsp70, which increases after experimental stroke. It transfers the death receptor Fas to the cell surface, inducing apoptosis after its ligands have bonded. A selective dynamin inhibitor also showed neuroprotective effects and may serve as a new therapeutic target. 46 Taken together, it seems that Hsp70 is mainly protective in stroke though detrimental effects such as the activation of the immune system.
It is the Y-box binding protein (YB-1), respectively DNA-binding protein B (DbpB), which is a prototype of the conserved cold shock domain proteins. It binds single-stranded RNA or DNA via two ribonucleoprotein domains (RNP-1 and RNP-2) to prevent annealing upon the induction of cold shock, and thus affects multiple cellular functions. 47 YB-1 takes part in a set of bacterial and sterile inflammatory processes.47,48 It is upregulated in proliferating microglia in an experimental ischemia model. 49 The cold-inducible RNA-binding protein (CIRP) also belongs to the group of stress-responsive proteins with RNPs. When it is released from microglia after experimental ischemic brain injury, it fosters neuroinflammation via CIRP-NFκB-mediated TNFα expression. 50 Moreover, in CIRP-deficient mice, the brain infarct volume, TNFα levels and activation of microglia are decreased. 51 Therapeutic hypothermia is discussed to be neuroprotective after stroke. 52 In experimental stroke research, hypothermia upregulates CIRP and Hsp70.1 and decreases TNFα and IL-6. 53 To sum up, both CIRP and YB-1 appear to have damaging functions in stroke.
Peroxiredoxins
Peroxiredoxin is a redox-active cytosolic DAMP, which is neuroprotective within cells by eliminating ROS, but loses its antioxidant capacity once released. 54 The oxidized peroxiredoxin 2 (PRDX2) is a set free from macrophages under inflammatory conditions and stimulates the production of TNFα. 55 Peroxiredoxin family proteins (Prxs) like Prx-1, Prx-2, Prx-5 and Prx-6 are released from necrotic brain cells 12 h after stroke onset and induce the production of proinflammatory cytokines like IL-23 from macrophages through TLR2 and TLR4 binding. 54 IL-23 induces IL-17-producing γδT cells and is responsible for delayed brain damage as neutrophils are recruited.56,57 Concordantly, the application of anti-IL-17 antibodies reduces infarct sizes and improves neurological outcome.58,59 Prx antibodies can reduce brain damage up to 12 h after ischemia. More than HMGB1, Prxs activate infiltrating macrophages. 54 Only exogenous Prx-1, Prx-2 and Prx-4 seem to act as proinflammatory in the ischemic brain through TLR4/NFκB signaling whereas Prx-3, Prx-5 and Prx-6 have no relevant effect on the production of NO, TNFα, IL-6, iNOS and TLR4 expression in macrophages. 60 In humans, low plasma levels of Prx-5 but high levels of Prx-1 during the first hours after stroke onset are found.61,62 Treatment with a gastrodin derivative, a constituent from the medicinal plant Gastrodiae rhizoma, reduced the post-ischemic expression of Prx-1, Prx-2 and Prx-4 after middle cerebral artery occlusion (MCAO) and the overall inflammatory response. 63 However, there is also evidence that Prx-2 can have neuroprotective effects in stroke as it inhibits both the poly (ADP-ribose) polymerase 1 (PARP1) and the p53-Bax-mediated death pathway. 64 Overall, peroxiredoxins mainly have damaging effects after stroke onset.
Purines
Extracellular nucleotides such as ATP serve as a multifunctional intercellular signaling system. Not only are they actively secreted through plasma membrane proteins such as pannexin and connexin but also passively released after stress-induced cell injury. 65 Ectonucleotidases such as CD39, CD73 and nucleoside diphosphate kinase degrade extracellular nucleotides in a stepwise process, eventually leading to adenosine. Extracellular nucleotides bind to the family of P2 receptors, which comprise both ligand-gated P2X cation channels and G-protein coupled P2Y receptors. 65 In the first 20 min after in vivo ischemia, the ATP extracellular concentration rises. Simultaneously, the degradation of ATP by ectonucleotidases is initiated so extracellular adenosine increases. This excitotoxicity returns to its basal level after about 4 h. 66 ATP concentration rises due to various mechanisms, for example, a vesicular release from neurons, exocytosis from astrocytes and microglia, or through membrane channels from glial cells and pyramidal neurons. 66 ATP itself aggravates cerebral damage after ischemic stroke as high ATP concentrations induce cell death. 67 All P1 and P2 receptor subtypes are expressed in neurons and glial cells, but also in lymphocytes and neutrophils. 66 Binding of the receptor P2X7 stimulates the inflammasome and enhances caspase-1 activity and the release of proinflammatory cytokines from innate immune cells. The receptor activation happens on K+ efflux through the cell membrane. 68 The highest concentrations of the purinergic receptor P2X7 can be measured on activated microglia, which leads to IL-1β release. The P2Y2 receptor is upregulated due to a rising IL1-β concentration after the P2X7 receptor is stimulated on microglia and astrocytes. 68 CD39-deficient mice, leading to less ATP metabolization, show larger infarct areas and more neutrophils and macrophages enter the CNS. 69 Hyman and colleagues have shown that CD39 on endothelial cells and leukocytes regulates their influx in the cerebral infarct zone. 70 The usage of a P2X7 antagonist or P2X7R blockade by Brilliant Blue G leads to a decrease in cerebral damage after ischemia in vivo.71–73 In contrast, Masuch and colleagues demonstrated a neuroprotective effect of microglia in organotypic hippocampal slice cultures, which is mediated by the P2X7 activation and the induction auf TNFα. 74 P2X7 activation also leads to alterations of the T-cell surface and can even cause T-cell death in the case of prolonged stimulation. 75 In contrast to ATP, adenosine, the end product of CD39, is considered to be neuroprotective. 66 Gaudin and colleagues created nanoparticles conjugated with adenosine, so that its metabolism was prolonged. As a result, decreased parenchymal damage after experimental cerebral ischemia was observed. 76 Concerning CD39, pretreatment with the small molecule inhibitor 3-(2,4-dichloro-5-methoxyphenyl)-2-sulfanyl-4(3H)-quinazolinone (Mdivi-1) raises adenosine in a MCAO mouse model, consequently protecting against cerebral ischemia. CD39 level was also increased as well as the cAMP response element-binding protein (CREB), which regulates CD39 expression. 77 In summary, purines are involved in the cellular reactions after ischemic stroke and it appears that ATP has a detrimental function whereas adenosine shows positive neuroprotective effects.
Other DAMPs in stroke
Apart from the discussed DAMPs, several more candidates have to be considered, including lipids and other proteins (S100A9; Mrp 8, Mrp9, CIRP, ASC specks). 78 Controversial opinions exist on the exact functions of these molecules in the field of stroke (for a review see Shichita and colleagues 78 ). In primary microglial culture, exogenous S100B appears to induce the expression of IL-1β, TNFα and iNOS, and in vivo injections of S100B activate microglia. These effects can be suppressed by the addition of poly (ADP-ribose) polymerase 1 (PARP-1). 79 Moreover, levels of hyaluronan, a large glycosaminoglycan in the extracellular matrix, are increased in patients with acute stroke. Concerning patients with intracerebral hemorrhagia, serum hyaluronan levels can be used for a prediction of the 3-month functional outcome 80 Cerebral hyaluronan expression is also induced by cortical ischemia, as shown in an experimental stroke mouse model. 81 Other endogenous TLR ligands, such as fibronectin, defensin or heparin sulfate proteoglycan, also activate TLR2 and TLR4, and are potentially involved in the initiation of sterile inflammatory responses. 82 Their role in stroke is still to be revealed. As a conclusion, various intracellular and also extracellular components might be involved as DAMPs in the inflammatory reactions after stroke, but until now their functions remain unclear.
DAMPs and microglia
Microglia are the resident immune cells of the cerebral nervous system and they are mainly responsible for the local immune surveillance.83,84 Overall, microglia is a heterogeneous population with a broad variety of functions and highly plastic. 85 Recently, a microglia-focused multicolor reporter mouse model monitored microglia dynamics in different states of health, disease and recovery. A self-renewing network with regional differences was described as being capable of reacting to different environmental challenges. Selected clonal expansion was observed in response to acute neurodegeneration as well as selected apoptosis in means of restoring homeostasis. 86 Neuronal cell injury alters the local microglia from a ramified phenotype to a macrophage-like one. 87
Regarding brain ischemia, hypoxia affects local microglia, which changes either into a proinflammatory or anti-inflammatory phenotype characterized by different secreted mediators. 88 There is no uniform response to injury as the relationship of microglia is complex. 89 More precisely, a spatiotemporal relationship between microglia morphology and localization of ischemic injury is established. 84 The activation of microglia is one of the first main responses to brain ischemia but continues for several weeks.6,88 In in vitro stroke models it was shown that microglia can worsen neuronal damage. 90 On the contrary, Szalay and colleagues elucidated thee protective effects. In an experimental study the authors treated their animals with the colony-stimulating factor 1 receptor CSF1R antagonist PLX3397 for 21 days, which resulted in a 98% decrease of microglia. This selective elimination of microglia led to a 60% augmented neuronal death after MCAO in mice. 91 Consequently, microglia are not yet completely understood as players in the concert of cellular reactions after ischemic stroke.
DAMPs interact with microglia and specific pathways are activated accounting for the polarization of microglia and their response to cell injury after stroke. One of the involved DAMPs is HMGB1. An autocrine loop includes the binding of glucocorticoids to microglia, which results in the release of the disulfide form of HMGB1 (dsHMGB1) and subsequent binding of dsHMGB1 to TLR2/4 as part of the priming process 92 [Figure 1(a)]. Inescapable tail shocks (ISs) in rat lead to elevated levels of HMGB1, an upregulation of the transcription factor NFκB and its NLRP3 inflammasome in microglia. 93 A second immune challenge results in a potentiated proinflammatory response of microglia with elevated levels of IL-1β. 94 Here, it is again the disulfide form of HMGB1, which induces the priming process. 22 Moreover, high concentrations of dsHMGB1 can be measured in the serum of patients with stroke. 92 When it comes to future therapeutic targeting of HMGB1, inflachromene inhibits microglia activation, and thus the release of HMGB1 with a reduced proinflammatory response. 95 As a result, HMGB1 is a main activator of microglia after ischemic stroke and its inhibition could possibly be a neuroprotective therapeutic option.
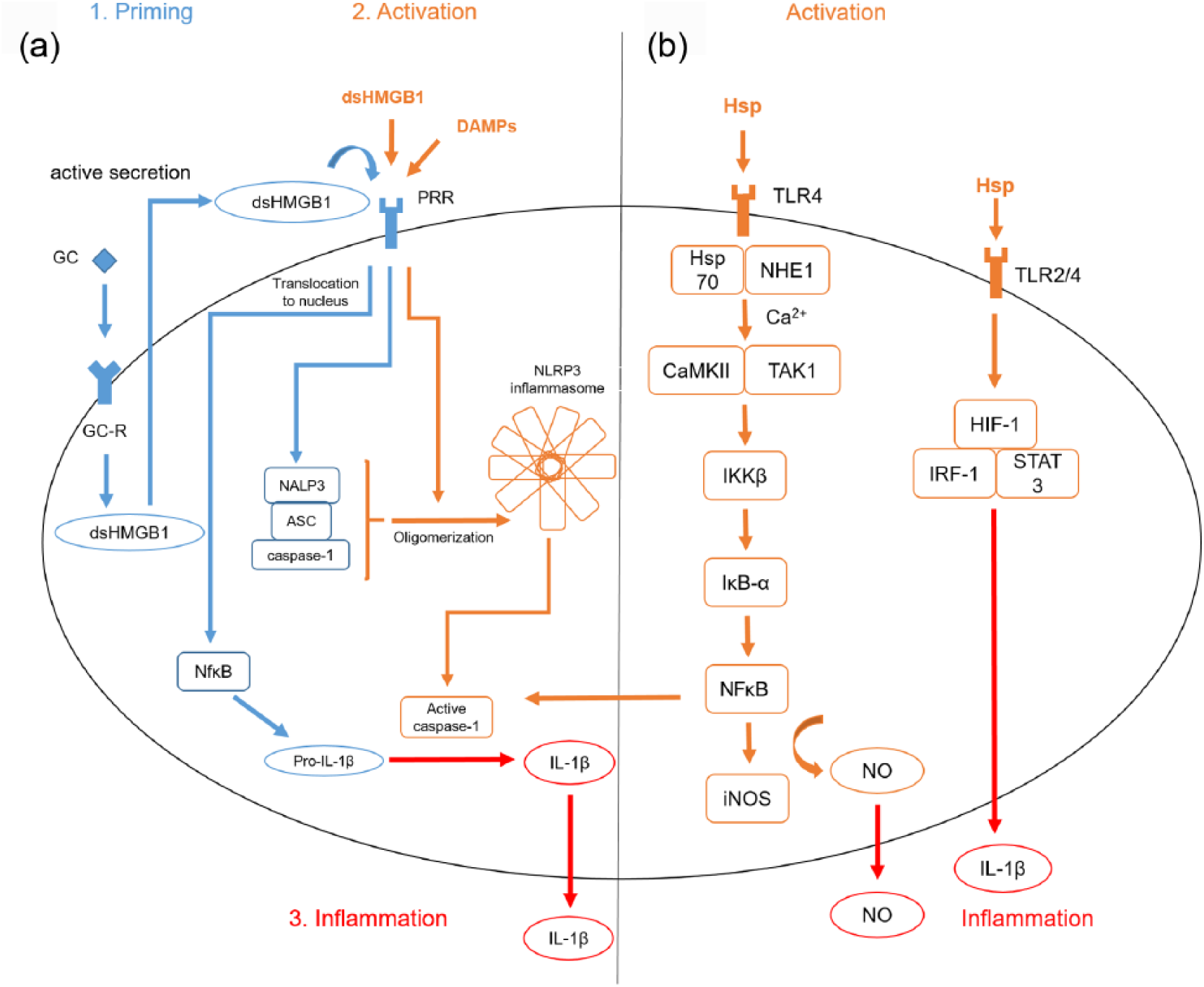
DAMP associated pathways in microglia.
Hsps also interact with microglia in stroke. In the case of danger, Hsp70 associates with the Na+/H+ exchanger 1 (NHE1), Ca2+ streams into the cell, which activates both the Ca2+-cal-modulin-dependent protein kinase II (CaMKII) and transforming growth factor β-activated kinase 1 (TAK1). IκB kinase β (IKKβ) initiates the degradation of IκB-α, and thus liberates NFκB resulting in a higher gene transcription of, for example, iNOS 97 [Figure 1(b)]. Whereas extracellularly located, Hsp activates microglia through TLRs, upregulating NFκB, hypoxia inducible factor 1 (HIF-1), interferon regulator factor 1 (IFR1) and single transducer and activator of transcription 3 (STAT3). This results in the release of proinflammatory cytokines such as IL-1β, IL-6, IL-12 and TNFα.41,42,98 Another player in the fields of microglia activation after stroke is Hsp72. It is the major cytosolic form of the 70 kDa family of Hsps and its overexpression in microglia is described to be cytoprotective as it interferes with the IKK-IκB-NFκB pathway.99,100 Doeppner and colleagues fused Hsp70 to the transactivator of transcription (TAT) domain of the human immunodeficiency virus to pass the blood–brain barrier. Intravenous infusion of TAT-Hsp70 reduced post-ischemic brain injury among others by diminished microglial activation. 44 Notably, Hsp90 inhibits 70 kDa inducible Hsp70 synthesis. This is why treatment with an Hsp90 inhibitor such as geldanamycin prevents lipopolysaccharide (LPS)-induced BV2-microglial cell death through subsequent Hsp70 induction. 101
Regarding peroxiredoxins, Prx-1 is an antioxidant mainly expressed in microglia that diminishes microglial activation in proinflammatory processes. 102 LPS is a randomly used inflammogen (PAMP) activating microglia in several mammalian model systems. TLR agonists such as LPS upregulate Prx-1 via a ROS/p38 mitogen-activated protein kinase (MAPK)-dependent pathway and suppresses NFκB signaling, and thus nitric oxide (NO) production 103 (Figure 2). The Nox (membranous nicotinamide adenine dinucleotide phosphate (NADPH) oxidase)/ROS (reactive oxygen species)/JNK (c-jun N-terminal kinase) axis is also activated by LPS, and hence, NO as an inflammatory mediator rises. Prx-5 is upregulated by LPS and lowers intracellular ROS levels in microglia. Thereby, a balanced inflammatory response is generated. 104 Furthermore, Prx-5 also reduces NFκB and MAPK activation in microglia after LPS application. 105 When it comes to sterile inflammation after stroke, Prx-3 was significantly elevated from 3 days post ischemia in an ischemia/reperfusion stroke model (I/R). As a result, phagocytic processes in the subacute phase after stroke can be assumed. 106 Prx-6 in microglia protects neurons against oxygen glucose deprivation and reoxygenation (OGD/R), showing a higher neuron viability. 107 Exogenous intraventricular Prx-3 treatment decreases microglia activation after experimental cerebral ischemia induction in gerbils. 106 Furthermore, spinal cord injury in rat increases Prx-1 expression in astrocytes and microglia, which also enhances microglia proliferation after inflammation. 108 Prx-1 was also induced in surrounding microglia after hemorrhagic stroke in rat. 109 Interestingly, it appears that microglia is not the target of Prxs but infiltrating immune cells releasing IL-17 and IL-23. 110 As a promising therapeutic approach, the natural compound obovatol suppresses Prx-2-mediated microglial activation in a NFκB-dependent manner. 111

Peroxiredoxin-associated pathways in microglia. Damaged-associated molecular patterns (DAMPs) such as peroxiredoxin family proteins Prx-1, -2 and -4 activate the the Nox (membranous NADPH oxidase) /ROS (reactive oxygen species)/ JNK (c-jun N-terminal kinase) axis and the concentrations of Nuclear-Factor κB (NFκB), mitogen-activated protein kinase (P38/MAPK) and reactive oxygen species (ROS) increase. Both Prx-1 and Prx-5 are upregulated and diminish the proinflammatory response by lowering ROS and NFκB signaling. Described pathways are best demonstrated by toll-like receptor (TLR) agonist lipopolysaccharide (LPS).
ATP is a major modulator of microglial function with at least four different expressed ATP receptors (P2X4, P2X7, P2Y6, P2Y12) (Figure 3). P2X7 activation through ATP in cultured mouse primary microglia results in an increased release of IL-6, CCL2 and TNFα. 112 Moreover, priming of cultured microglia with TLR isoform ligands such as LPS, a TLR4 ligand, lead to a release of IL-1β upon ATP application. The receptors TLR2, -3 and -4 are all found on microglia. The ATP-dependent release of IL-1β after pretreatment with TLR agonists can be blocked by a P2X7 antagonist A7410003 in primary cultures of microglia. 113 Regarding the oxygen/glucose deprivation (OGD) model, an accepted cell culture stroke model, the ATP receptors P2X4 and P2X7 are upregulated on microglia, which accounts for cellular damage after OGD. Purinergic antagonists decreased cell injury under these conditions. 114 The P2Y6 receptor on microglia is activated by uridine diphosphate (UDP) from damaged neurons, causing microglial phagocytosis, and thus limiting the inflammatory response after cell injury in in vitro and in vivo rat models. 115 Phagocytosis is also enhanced by P2Y2/4 and P2Y12 receptors. 116 In contrast to that, the P2Y12 receptor on microglia has a proinflammatory and chemotactic effect in brain ischemia models as inhibition improves neuron viability, which is interesting. As the common use of the antiplateleta agent clopidogrel targets P2Y12, an additional therapeutic side effect can be assumed. 117 Moreover, P2Y-receptor-mediated Ca2+ signaling and migration of microglia, which are mainly dependent on P2Y1 and P2Y6 receptors, are altered by extracellular acidosis. 118 On the other hand, 2’,3’-cAMP released from damaged cells is metabolized by an extracellular pathway on microglia to 2’- and 3’-AMP, which is then metabolized to adenosine. Adenosine has a neuroprotective role if bound to its A1 receptor on microglia. Therefore, this 2’,3’-cAMP-adenosine pathway functions as a limiting factor to the microglial response after cell injury. 119 In LPS-stimulated microglia, a reduced number of A1 receptors can be observed whereby ATP limits its own activation potential. 120

Nucleotide-associated pathways in microglia. A toll-like receptor (TLR) agonist such as lipopolysaccharide (LPS) leads to the release of proinflammatory mediators such as interleukin (IL)-6 in an ATP-dependent manner. Signaling through P2X4 receptors enhances migration of microglia. Uridine triphosphate (UTP) binds to P2Y2/4, Uridine diphosphate (UDP) to P2Y6 and adenosine diphosphate (ADP) to P2Y12, which results in a higher rate of phagocytosis. 2’,3’-cyclic adenosine monophosphate (cAMP) is released from damaged cells and is metabolized by an extracellular pathway to 2’- and 3’-adenosine monophosphate (AMP), which is then metabolized to adenosine. After adenosine has activated its A1 receptor, it possesses neuroprotective functions.
In summary, DAMPs play a major role in microglia activation and possess both neuroprotective and inflammatory functions.
DAMPs and other cell types
Besides microglia, other brain resident cells can also be activated through DAMPs.
Concerning astrocytes, the expression of TLRs like TLR2 and -4 can be induced by DAMPs and other soluble factors, including interferon γ. 121 In an oxygen-glucose deprivation/reoxygenation (OGD/R) model, the pathologic swelling of astrocytes can be mediated through TLR4, myeloid differentiation primary response gene 88 (MyD88) and NFκB. 122 Further, it is known that astrocytes exhibit a PI3K/Akt-mediated upregulation of HMGB1 and IL-6 in the ischemic brain, which is crucial for their proangiogenic function. 123 Moreover, a feedback loop between microglia and astrocytes exists. Following activation of microglia in primary cocultures, microglia-derived ATP recruits astrocytes, which in turn are a potential source of neurotoxic glutamate. 124
Regarding endothelial cells, it has recently been shown that HMGB1 activates monocytes and endothelia via RAGE after stroke, resulting in atheroprogression, and thus vascular inflammation. 125 It is described that polarized peroxiredoxin 1 localizes to endothelial cells from ipsilateral brain microvessels 12 h after transient middle cerebral artery occlusion (tMCAO) induction in vivo, which might be involved in its neuroprotective effect on tight junction proteins. 126 Additionally, it has been shown that in the early phase after ischemia, peroxiredoxin 1 is upregulated and owns neuroprotective functions whereas in the later stages the degradation of peroxiredoxin 1, mediated by the E3 ubiquitin ligase E6-associated protein (E6AP), results in neurovascular damage. 127
When it comes to oligodendrocytes, an autocrine HMGB1-TLR2 axis is hypothesized. Following ischemia, released HMGB1 binds to TLR2, which results in the activation of protective signaling pathways. 128 In this context, HMGB1 acts as an endogenous trophic factor for oligodendrocytes, supporting white matter integrity following ischemia. 129
In summary, the role of DAMPs on other cell types is diverse and further attempts are necessary to elucidate the exact involvement in means of stroke.
Conclusions and perspectives
A set of DAMPs were outlined in this review. Besides the neuroprotective effects of cold domain proteins or adenosine, proinflammatory reactions were described as induced by ATP or HMGB1. Therefore, DAMPs possess a wide variety of functions with context-specific effects dependent on cell type or intra- versus extracellular location. As released by necrotic cells after brain ischemia, they take part in the early reactions after stroke onset. Both the innate and the innate immune system are substantial players in the cellular concert after ischemia. The list of DAMPs can be extended by numerous other protein DAMPs, such as S100, serum amyloid A, histones and nonprotein DAMPs such as heparin sulfate, DNA or RNA. 10 Their function is still to be further distinguished. Moreover, we shed light on the involvement of microglia, which play a major role in the acute innate immune reaction after brain ischemia. Microglia are affected by DAMPs but also secrete them actively. What is more, initial therapeutic strategies targeting DAMPs were discussed highlighting new methods in stroke therapy.
Footnotes
Funding
This work was supported by a grant from the Hermann and Lilly Schilling foundation.
Conflict of interest statement
The authors declare that there is no conflict of interest.