
Abstract
Cerebellar ataxias represent a group of heterogeneous disorders impacting on activities of daily living and quality of life. Various therapies have been proposed to improve symptoms in cerebellar ataxias. This review examines the physiological background of the various treatments currently administered worldwide. We analyze the mechanisms of action of drugs with a focus on aminopyridines and other antiataxic medications, of noninvasive cerebellar stimulation, and of motor rehabilitation. Considering the cerebellum as a controller, we propose the novel concept of ‘restorable stage’. Because of its unique anatomical architecture and its diffuse connectivity in particular with the cerebral cortex, keeping in mind the anatomophysiology of the cerebellar circuitry is a necessary step to understand the rationale of therapies of cerebellar ataxias and develop novel therapeutic tools.
Keywords
Introduction
Cerebellar ataxias (CAs) have originally been described as being characterized by motor incoordination [Manto, 2008; Bodranghien et al. 2016; Manto et al. 2012]. When persistent, CAs interfere significantly with daily life activities. Although various therapies are available for CAs, treatment of the underlying disease is available only for infarction/hemorrhage, metabolic/toxic disease, immune-mediated CAs (IMCAs) and some cerebellar malformations. In degenerative CAs, therapies are missing or provide only minimal benefits. One explanation for this important gap is that therapies of CAs have not been developed on the basis of the pathogenesis. A better understanding of the mechanisms involved in the pathophysiology of degenerative CAs should clearly help the design of new treatments [Ilg et al. 2014].
There is no doubt that even novel therapies will not completely abolish the clinical deficits associated with CAs, especially when a noticeable neuronal loss has already occurred. Acting on residual cerebellar function is thus an objective which deserves attention. In particular, neuromodulation therapies aim to reinforce residual cerebellar function and could thus be potentially appropriate when cerebellar function is impaired. Motor rehabilitation complements drug therapies in most cases. Recent studies report in particular on the efficacy of aminopyridines, acting as K+ channel blockers, and noninvasive cerebellar stimulation in CAs [Ilg et al. 2014]. Importantly, the mechanisms of action of neuromodulation therapies are based on physiological theories considering that the role of the cerebellum is to act as an adaptive modulator. This modulator is assumed to control timing of muscles discharges and synergy between muscles [Manto, 2008; Manto et al. 2012]. However, a consensus on an integrated hypothesis is currently lacking [Manto, 2008; Manto et al. 2012].
In this review, we examine the physiological background of each therapy. First, we briefly summarize the currently available treatments for each CA. An algorithm for the diagnosis and therapy of the most common CAs is proposed. We suggest the concept of ‘restorable stage’ in which an appropriate therapy can restore or augment cerebellar functions in the presence of residual cerebellar function. Finally, we discuss the pathophysiological mechanisms underlying the action of neuromodulation therapies. The efficacy of aminopyridine, other drugs, noninvasive cerebellar stimulation, and motor rehabilitation is discussed from the viewpoint of cerebellar motor control.
The aim of this study is mainly directed at a critical review of the impact of therapies on cerebellar motor function, with an emphasis on the physiology of the cerebellum. Possible treatment effects on cerebellar cognition and affect are beyond the scope of this review.
Presentation of the most common cerebellar ataxias
Differential diagnosis on the basis of the clinical presentation
Acute cerebellar ataxias
Figure 1 shows our algorithm for the differential diagnosis of acute CAs (ACAs). Cerebellar infarction, hemorrhage, Wernicke encephalopathy, ACA/acute cerebellitis (AC), immune-mediated CAs (IMCAs), or cerebellar injuries should be considered.
Cerebellar stroke. Although cerebellar stroke typically presents with a sudden onset, some patients do not report an acute onset and visit the hospital only a few days after the onset of symptoms, complaining mainly of dizziness, vertigo, or vomiting, or mild instability. Thus, the differential diagnosis of a gradual worsening of symptoms over a period of a few days should also include cerebellar stroke, in addition to Wernicke encephalopathy, ACA/AC and IMCAs.
ACA/AC. These terms are confusing. Based on the criteria listed by Sawaishi and Takada [Sawaishi and Takada, 2002], ACA is defined as a self-limiting disease caused by viral infection (especially chickenpox) or immunization-induced immune mechanisms in childhood, whereas AC is defined as a disease caused by direct invasion of a pathogenic agent.
IMCAs. In addition to Miller–Fisher syndrome, some patients with IMCAs (paraneoplastic cerebellar degeneration, ‘anti-glutamic acid decarboxylase (GAD) antibodies associated CA) exhibit an ACA. CAs with acute onset may also develop in patients with multiple sclerosis (MS), although cerebellar symptoms occurring in isolation are not common in this disorder.
Cerebellar injuries. Traumatic injury to the posterior fossa is a heterogeneous condition which may cause cerebellar hematoma, epidural hematoma or subdural hematoma. Symptoms of ataxia are masked by an impaired consciousness in many of patients. Posterior fossa trauma may be associated with a rapid neurological decline, which results in a high rate of morbidity compared with supratentorial trauma [Nixon et al. 2014]. Thus, the importance of early identification is stressed [Nixon et al. 2014].

Algorithm for the diagnosis and treatment of patients with acute cerebellar ataxias. ACA/AC, acute cerebellar ataxia/acute cerebellitis; CT, computed tomography; hemorrhage, cerebellar hemorrhage; infarction, cerebellar infarction; injuries, cerebellar injuries; IMCA, immune-mediated cerebellar ataxia; MRI, magnetic resonance imaging; MS, multiple sclerosis; rtPA, recombinant tissue plasmin activator; Wernicke, Wernicke encephalopathy.
Imaging studies [computed tomography (CT) and magnetic resonance imaging (MRI)] are used for the differential diagnosis of the above entities. In cerebellar stroke and cerebellitis, the swollen cerebellum sometimes compresses the brainstem or the fourth ventricle, resulting in obstruction of cerebrospinal fluid (CSF). Thus, the presence or absence of hydrocephalus should be carefully determined.
Subacute, chronic, or insidious cerebellar ataxias
Figure 2 shows our algorithm for the differential diagnosis of subacute, chronic, or insidious CAs. After checking for an exposure to certain toxic agents, such as ethanol, organic mercury, organic solvent (toluene, thinner), certain medications (e.g. phenytoin, metronidazole) and after excluding physical signs of hypothyroidism, imaging studies (e.g. MRI) are conducted to look for a displacement of the tonsil, a tumor, an inflammation or a cerebellar atrophy. In general, the presence of cerebellar atrophy is suggestive of degenerative CAs. The severity of cerebellar atrophy correlates with the degree of CAs. Further genetic analysis should be conducted for the differential diagnosis of autosomal dominant CAs (ADCAs) and autosomal recessive CAs. In the case of pure cerebellar atrophy or multiple systemic atrophy, CA is sporadic (although some cases of pure cerebellar atrophy may have a genetic origin). The presence of mild cerebellar atrophy relative to the clinical presentation of CAs is often indicative of IMCAs. Serological tests, including measurement of autoantibodies, should be performed for the diagnosis of the subtype of IMCAs.

Algorithm for the diagnosis and treatment of patients with subacute, chronic or insidious cerebellar ataxia (CA). Chiari, Chiari syndrome; IMCA, immune-mediated cerebellar ataxia; MRI, magnetic resonance imaging; MS, multiple sclerosis.
Malformations
Decompressive surgery (posterior fossa decompression, with or without spinal laminectomy) is necessary in patients with Chiari syndrome who present with compression of the brainstem by cerebellar tonsil displacement [Greenberg, 2006].
Vascular diseases
Cerebellar infarction
The infarct core is usually surrounded by the hypoxic area (i.e. ischemic penumbra). The ischemic penumbra contains electrically silent neurons that undergo cell death without salvage, and provides the rationale for immediate thrombolytic therapy [Astrup et al. 1981]. The standard therapy is intravenous injection of recombinant tissue plasmin activator within 4.5 h from the onset of infarction [Hacke et al. 2008]. However, there are two problems in the management of cerebellar infarcts. First, early diagnosis within the 4.5 h window is sometimes difficult, especially when the main complaints are dizziness, vertigo, and vomiting [Wijdicks et al. 2014]. Without careful attention to eye movements, speech, and walking instability, the chance for thrombolytic therapy might be missed. Second, poststroke cerebellar swelling can compress the brainstem sometimes with a delay, resulting in reduced conscious level and appearance of brainstem signs, such as loss of corneal reflex and miosis [Wijdicks et al. 2014]. A large retrospective study of 84 patients with extensive cerebellar infarction showed that 40% of these patients required craniotomies and 17% required ventricular drainage [Jauss et al. 1992]. After craniotomies and drainage, 74% of these patients showed good outcome [Jauss et al. 1992]. Suboccipital craniotomy with dural expansion is recommended for cerebellar infarcts with wide areas of edema [Wijdicks et al. 2014].
Cerebellar hemorrhage
Some patients with cerebellar hemorrhage are admitted in a state of coma, while others show deterioration of consciousness a few days after the onset [Jensen and St Louis, 2005]. The main features that predict clinical deterioration include systolic blood pressure at admission of over 200 mmHg, pinpoint pupils, abnormal corneal or occulocephalic reflexes, extension of a hemorrhage into the vermis, a hematoma greater than 3 cm, visible brainstem compression, presence of intraventricular hemorrhage, upward herniation, and acute hydrocephalus [Jensen and St Louis, 2005]. In 1999, the American Stroke Association Guidelines recommended surgical removal of the hemorrhage in patients with a hematoma measuring over 3 cm in size in case of neurological deterioration, and also in patients with brainstem compression or hydrocephalus caused by ventricular obstruction. The size requirement of the hematoma (i.e. >3 cm) was abandoned in the 2010 revised guidelines [Morgenstern et al. 2010].
Common problems underlying therapies for cerebellar stroke
The main purpose of cerebellar stroke therapy is to avoid damage to neighboring normal structures surrounding the site of infarction or hemorrhage, such as the ischemic penumbra or brainstem, so as to preserve the maximal potential for reversibility. In particular, the expanding edema or hematoma can potentially compress brainstem tracts or the fourth ventricle, the obstruction of which causes acute hydrocephalus. Both conditions can lead to coma and death. Thus, repeated neurological examination and CT or MRI are also recommended within the 72–96 h, post stroke period [Wijdicks et al. 2014].
Metabolic and toxic diseases
Hypothyroidism, Wernicke encephalopathy, Niemann-Pick disease type C, and cerebrotendinous xanthomatosis
In the case of hypothyroidism-induced CA, thyroid hormone (thyroxine) is replenished [Rowland, 2005]. Wernicke encephalopathy may or may not be related to alcohol abuse [Scalzo et al. 2015]. It is widely accepted that clinical suspicion of vitamin B1 deficiency requires an immediate treatment with vitamin B1 [Rowland, 2005; Scalzo et al. 2015]. Whole blood sample should be taken before the commencement of such therapy for definite diagnosis.
Niemann-Pick disease type C is a rare disorder characterized by mutations in NPC1 or 2 gene, leading to an intracellular lipid-trafficking deficit with secondary accumulation of glycosphingolipids [Patterson et al. 2007]. A randomized controlled study showed that miglustat, an inhibitor of glucosylceramide synthesis, improves saccadic eye movements, swallowing capacity, and ambulatory capacity [Patterson et al. 2007]. Recently, 2-hydroxypropyl-beta-cyclodextrins (HPBCD) has gained attention as a promising candidate [Camargo et al. 2001; Nusca et al. 2014; Vite et al. 2015]. HPBCD increases the solubility of poorly water-soluble compounds such as cholesterol [Vite et al. 2015]. Intrathecal administration of HPBCD reduces ataxic symptoms and cell loss in affected cats [Vite et al. 2015].
Cerebrotendinous xanthomatosis is another rare disease caused by mutations in the CYP27A1 gene, a converting enzyme of cholesterol to cholic and chenodeoxycholic acids [Keren and Falik-Zaccai, 2009; Nie et al. 2014]. The reduced level of cholic and chenodeoxycholic acids elicits, in turn, a lack of cholesterol synthesis, resulting in a high level of serum cholestanol and accumulation of lipids in the central nervous system and connective tissues [Keren and Falik-Zaccai, 2009]. It is established that replacement therapy using chenodeoxycholic acid improves neurological symptoms and contributes to a better prognosis [Nie et al. 2014]. Early diagnosis and long-term treatment are recommended in these two autosomal recessive inherited diseases [Nie et al. 2014].
Toxic diseases
Exposure to toxic agents, such as ethanol, organic mercury, organic solvent (toluene, thinner) and certain medications (e.g. phenytoin, metronidazole) can induce CAs. The first line of therapy is to remove the causative toxic agent to prevent clinical worsening or cerebellar atrophy [Rowland, 2005].
Neoplasms
Primary neoplasms causing CAs can be classified into two groups: tumors that grow within the cerebellum and tumors that grow outside the cerebellum (mainly the cerebellopontine angle) with compression of the cerebellum (Table 1) [Greenburg, 2006]. Low-grade tumors [hemangioblastoma, pilocytic astrocytoma, vestibular schwannoma, meningioma, and epidermoid cyst; World Health Organization (WHO) grade I], with no invasion of the cerebellar tissue, can be surgically excised. However, ependymoma (WHO grade II or III) requires surgical excision followed sometimes by radiotherapy [Greenburg, 2006]. Primary high-grade tumors (medulloblastoma; WHO grade IV) also require the combination of surgery and radiotherapy, and even chemotherapy, especially in high-risk cases [Greenburg, 2006].
Treatment of neoplasms within or surrounding the cerebellum.

WHO, World Health Organization.
In contrast, surgery is difficult in metastatic tumors, except in cases of solitary tumors, especially since chemotherapy is usually ineffective. Thus, the first-line treatment is radiotherapy [Greenburg, 2006].
Acute cerebellar ataxia and acute cerebellitis
ACA usually follows a self-limited benign course without complications. In contrast, AC is potentially associated with cerebellar swelling, which may result in compression of the brainstem or obstructive hydrocephalus [Sawaishi and Takada, 2002]. Various bacteria and viruses are known to cause AC, including Epstein–Barr virus (EBV), Herpes simplex virus (HSV), Varicella zoster virus (VZV), respiratory syncytial (RS) virus, Coxsackie B3 virus, rubella virus, Listeria, Mycoplasma pneumoniae, and Coxiella burnetti [Sawaishi and Takada, 2002], although no microorganism is identified in some cases [Sawaishi and Takada, 2002]. AC is caused by direct invasion of specific microorganisms and requires immediate treatment with antiviral drugs (e.g. acyclovir for HSV and VZV) or antibiotics (e.g. ampicillin for Listeria, minocycline for Coxiella burnetti) [Sawaishi and Takada, 2002].
Deterioration of consciousness accompanied by diffuse cerebellar swelling should be treated immediately with mannitol or glycerol and corticosteroids [Sawaishi and Takada, 2002]. Development of obstructive hydrocephalus requires immediate surgical treatment (ventricular drainage and/or suboccipital craniotomy with dural expansion) [Sawaishi and Takada, 2002].
Immune-mediated cerebellar ataxias
MS is a representative disease of IMCAs. The standard treatment for MS is an induction therapy using corticosteroids, followed by maintenance therapy using interferon β [Weier et al. 2015]. Interestingly, new guidelines for the treatment of other IMCAs have been proposed recently [Mitoma et al. 2015, 2016]. This category includes gluten ataxia, paraneoplastic cerebellar degeneration, anti-GAD antibodies associated CA, and steroid-response IMCA associated with antithyroid antibodies. These diseases can be differentiated by their clinical course and specific associated autoantibodies. When CAs are triggered by certain known antigenic stimulants, priority should be given to treatment of the underlying condition [Mitoma et al. 2015]. For example, gluten-free diet in gluten ataxia and surgical removal of the neoplasm in paraneoplastic cerebellar degeneration. Progression of CAs after treatment of the underlying condition requires immediate immunotherapy. However, when CAs are not triggered by any other condition (e.g. anti-GAD antibodies associated CA, and steroid-response IMCA associated with antithyroid antibodies), induction immunotherapy is provided first to minimize CAs, followed by maintenance immunotherapy to prevent relapse [Mitoma et al. 2015]. Various combinations of immunotherapies have been used, ranging from intravenous immunoglobulins, corticosteroids, immunosuppressants, plasmapheresis, and rituximab, depending on the subtype of IMCA (Table 2) [Mitoma et al. 2015].
First-line immunotherapy for each subtype of immune-mediated cerebellar ataxia.
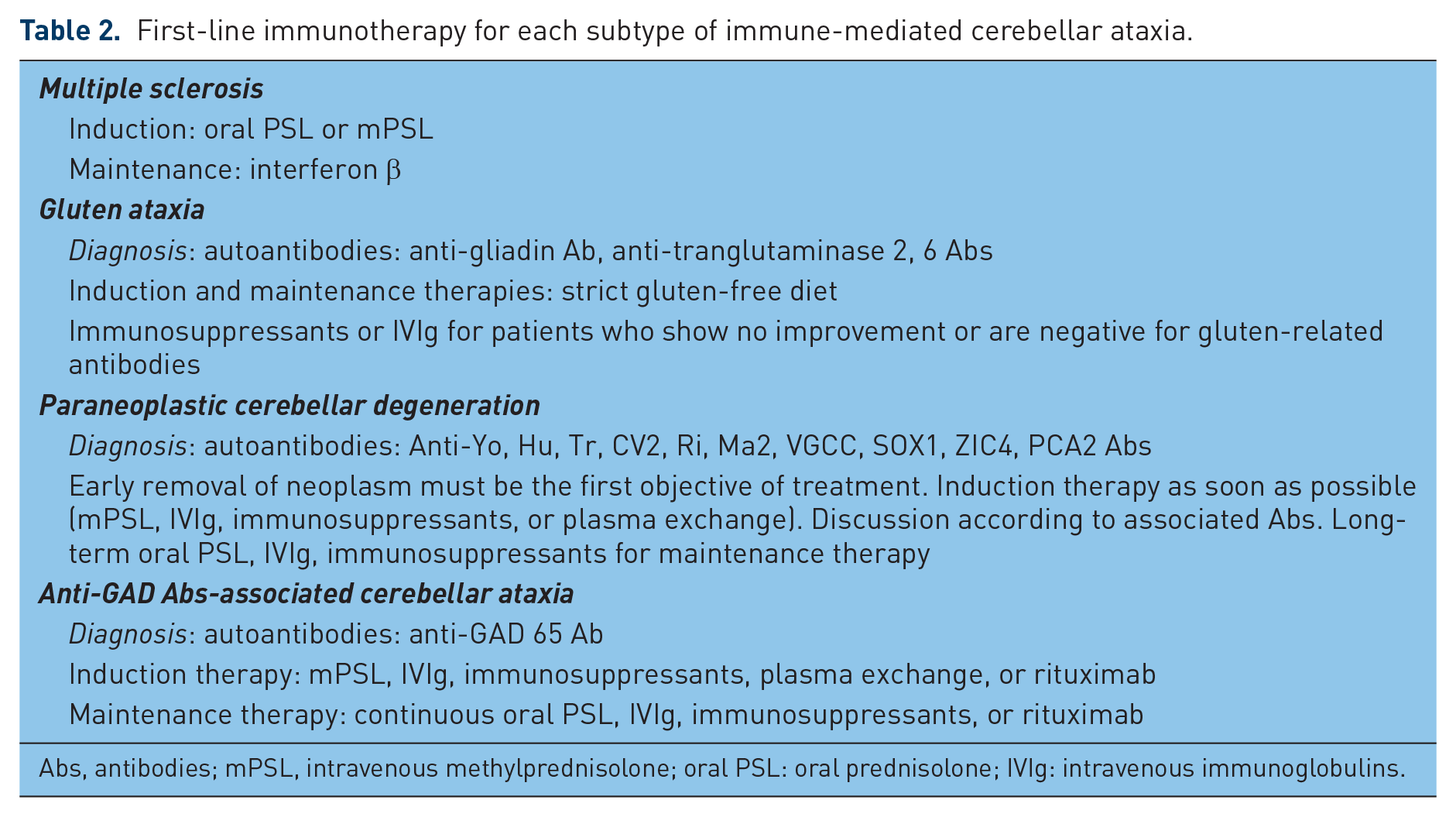
Abs, antibodies; mPSL, intravenous methylprednisolone; oral PSL: oral prednisolone; IVIg: intravenous immunoglobulins.
Degenerative cerebellar ataxias
Friedreich’s ataxia, a representative sensory ataxia, is caused by lack of frataxin. This mitochondrial protein is involved in iron-sulfur cluster synthesis [Stemmler et al. 2010], which provides clues for therapy [Ilg et al. 2014]. Ataxia with isolated vitamin E deficiency is characterized by a deficit in α-tocopherol transfer protein (a protein involved in intracellular transport of α tocopherol). Patients develop sensory ataxia with loss of propioception and retinitis pigmentosa [Yokota et al. 1997]. Oral supplementation of vitamin E was reported to halt the progression of neurological symptoms and retinitis pigmentosa [Yokota et al. 1997].
In contrast to sensory ataxias, therapies directed against the cause of CAs have been disappointing in pure cerebellar degeneration [Ilg et al. 2014]. However, one therapeutic approach could be effective in the Cuban type of spinocerebellar ataxia type 2 (SCA2), which is associated with low serum and CSF zinc concentrations [Velázquez-Pérez et al. 2011]. In this regard, a double-blind, placebo-controlled study shows that ZnSO4 supplementation increased Zn level in the CSF and resulted in a simultaneous improvement of the subscore for gait, posture, and alternating hand movements as well as a reduction of saccadic latency as compared with placebo on the Scale of Assessment and Rating of Ataxia (SARA) [Velázquez-Pérez et al. 2011]. Based on these findings, checking plasma and CSF zinc concentrations is recommended in cases with pure cerebellar degeneration [Ilg et al. 2014].
Cerebellar injuries
Due to the progressive and worse prognosis, nonsurgical management should be considered only when patients are fully consciousness and posterior fossa lesions are small with little or no mass effect [Nixon et al. 2014]. Immediate surgical removal of hematoma is recommended in the case of an epidural hematoma greater than 30 cm3, a subdural hematoma greater than 10 mm, or an obstruction or compression of the fourth ventricle [Nixon et al. 2014].
Prospective problems in treatment directed against the underlying disease
In contrast to CAs associated with vascular, toxic or metabolic diseases, and AC, certain subtypes of IMCAs (paraneoplastic cerebellar degenerations and chronic type of anti-GAD antibodies associated CA) and degenerative CAs cannot be prevented. However, new types of autoantibodies that target intracellular signal transduction have been discovered [Mitoma et al. 2015]. Details of the mechanisms of cell death in degenerative CAs have also been published in recent years [Ilg et al. 2014]. These findings could provide new clues to keep cerebellar damage at a minimum level in each disease.
Interestingly, a cross correlation of neurodegeneration with immune deficiency has been recently reported. In addition, interposed nucleus neurons may modulate the immune system via the hypothalamus [Lu et al. 2015]. As a result, degeneration of the nuclear neurons may have a direct impact on the immune system.
Neuromodulation therapy using aminopyridines
The K+ channel blockers aminopyridines [4-aminopyridine (4-AP), 3, 4-diaminopyridine (3, 4-DAP)], have been used in patients with episodic ataxia 2 (EA2), in patients exhibiting a downbeat nystagmus (DBN), and in patients with other degenerative CAs.
Episodic ataxia 2
EA2 is characterized by attacks of ataxia [Jen et al. 2007]. Acetazolamide (a carbonic anhydrase inhibitor) decreases or attenuates such attacks in 50–70% of the patients [Strupp et al. 2011]. However, this effect was later found to wane with time in patients on long-term treatment [Strupp et al. 2011]. Furthermore, the adverse effects of acetazolamide (renal tubular acidosis with nephrolithiasis, hyperhidrosis, paresthesia, and muscle stiffening) also limit its clinical use [Strupp et al. 2011].
Clinical evidence
Several case reports and retrospective studies have shown that 4-AP prevents CA attacks [Strupp et al. 2004, 2008; Claassen et al. 2013]. Strupp and colleagues conducted a randomized, double-blind, and crossover clinical trial of 10 patients treated with EA2 and provided convincing evidence for the efficacy of 4-AP [Strupp et al. 2011]. Before treatment, the patients presented with episodic ocular ataxic symptoms, sometimes accompanied by limb ataxia and ataxic gaits. Treatment with 4-AP (5 mg three times daily) significantly reduced the frequency of episodic CAs (placebo: 6.5 attacks/month; 4-AP: 1.65 attacks/month, p = 0.03). Furthermore, 4-AP tended to reduce the duration of the attack (from 13.65 h after placebo to 4.45 h after 4-AP), although the difference was not statistically significant (p = 0.08). 4-AP also significantly reduces the severity of attacks, as determined by the Vestibular Disorders Activities of Daily Living Scale.
Mechanisms of action
Aminopyridines do not improve cerebellar synaptic transmission
A deficit in P/Q-type Ca2+ channels was previously assumed to impair cerebellar synaptic transmission, leading to the development of CAs [Alvina and Khodakhah, 2010]. Specifically, it was thought that a decrease in P/Q-type Ca2+ current reduces the inhibition of deep cerebellar nuclei (DCN) in EA2 and that aminopyridines could restore the depressive input on DCN and thus improve CAs [Glasauer et al. 2005]. Although changes in cerebellar synaptic transmission were evident in some animal models of EA2, they are relatively minor, presumably due to compensation by other Ca2+ channels [Matsushita et al. 2002; Ovsepian and Friel, 2008]. Alvina and Khodakhah also demonstrated that at therapeutic concentrations, 4-AP did not increase the spontaneous activities of Purkinje cells (PCs) or the release probability at parallel fiber–PC and PC–DCN neuron synapses [Alvina and Khodakhah, 2010].
Aminopyridines restore distorted precision of PC pacemaking
The P/Q-type Ca2+ current is coupled with activation of KCa channels [Womack et al. 2004]. However, KCa channels limit the maximum PC firing rate by contributing to slow afterhyperpolarization, which plays a crucial role in the regulation of spontaneous firing [Womack and Khodakhaha, 2003]. Thus, in P/Q-type voltage-gated Ca2+ channel mutations associated with EA2, impaired precision of intrinsic PC pacemaking is expected. This assumption was clearly demonstrated in experiments involving ataxic mutation mice [Walter et al. 2006]. Distorted pacemaking was associated with lesser activation of KCa with each action potential, leading to a lower amplitude afterhyperpolarization and reduced potassium conductance during interspike intervals. In addition, the authors reported that 1-ethyl-2-benzimidazolinone (EBIO), an activator of KCa channels, restored the regularity of PC firing, resulting in improvement of ataxic movements in mutant mice.
Alvina and Khodakhah demonstrated that therapeutic concentrations of 4-AP blocked the Kv1 family of K+ channels, leading to a prolongation of the duration of action potentials and an increase in afterhyperpolarization, which resulted in restoration of pacemaking precision in PCs of mice with P/Q-type Ca2+ channel mutations [Alvina and Khodakhah, 2010]. The mechanisms of action of 4-AP are currently explained as follows: 4-AP-induced blockade of the Kv1 family of K+ channels widens action potentials; the broader action potential induces larger Ca2+ influx, which compensates the reduced P/Q-type Ca2+ current density associated with EA2 mutation; and restored Ca2+ currents result in additional activation of Ca2+-dependent K+ conductance [Alvina and Khodakhah, 2010]. Importantly, 4-AP-induced restoration of pacemaking precision correlated with improvement of ataxic movements in P/Q-type Ca2+ channel mutant mice [Alvina and Khodakhah, 2010].
Restoration of PC pacemaking could improve CAs
Timing is essential for the production of rapid movements and coordination of dynamic interaction in multijoint movements [Manto et al. 2012; Ivry, 1997]. Certain ataxic signs are due to a deficit in the control of timing [Manto, 2008; Manto et al. 2012]. The physiological assumption that the cerebellum acts as a time control machine originates from Braintenberg who compared the cerebellar cortex to a clock with a system of delay lines (corresponding anatomically to parallel fibers) [Braitenberg and Atwood, 1958]. Llinās proposed that rhythmic activities of the inferior olive nucleus synchronize a group of PC (via the climbing fibers) so as to fine tune timing (see Figure 3) [Llinas, 2009]. Recently, a temporal processing within the cerebellum has also been proposed [Manto et al. 2012; Ivry, 1997].

A scheme of cerebellar neural circuits.
A series of studies on EA2 and aminopyridines showed that the accuracy of pacemaking in PCs correlated with the capacity of coordination in P/Q-type voltage-gated Ca2+ channel mutation mice [Alvina and Khodakhah, 2010; Walter et al. 2006]. The correlation between the accuracy of PC pacemaking and coordination could be explained by the notion of temporal processing. Thus, it is argued that a distorted PC pacemaking, which potentially serves as an inappropriate clock [Walter et al. 2006], might impair cerebellar timing control so as to result in CAs, although the exact mechanism in the cerebellar circuitry remains unclear. Clinically, these studies have proved that the function of PC pacemaking can be a therapeutic target in EA2.
Downbeat nystagmus
DBN is the most frequent form of acquired persistent fixation nystagmus [Baloh and Spooner, 1981; Pierrot-Deseilligny and Milea, 2005]. It consists of two phases: a spontaneous upward drift and a fast downward phase during gaze straight ahead [Baloh and Spooner, 1981; Pierrot-Deseilligny and Milea, 2005]. DBN occurs not only in patients with degenerative CAs, but also in those with infarction, Chiari syndrome, MS, and IMCAs [Baloh and Spooner, 1981; Pierrot-Deseilligny and Milea, 2005; Kalla et al. 2007].
Clinical evidence
Aminopyridines have been reported to improve DBN in patients with various pathologies [Kalla et al. 2007; Strupp et al. 2003; Kalla et al. 2004]. Strupp and colleagues conducted a placebo-controlled, double-blind study to examine the effects of 3, 4-DAP (20 mg/day) on DBN in 17 patients, including five patients with degenerative CAs, three patients with infarction, one patient with Arnold–Chiari syndrome, and eight patients with idiopathic DBN [Strupp et al. 2003]. They reported that 3, 4-DAP reduced the mean slow phase velocity from 7.2 ± 4.2° to 3.5 ± 2.5° in DBN. Kalla and colleagues examined the effects of 4-AP (at a dose of up to 50 mg/day) in 15 patients with DBN (four patients with degenerative CAs, five patients with idiopathic DBN, and six patients with other etiologies) and healthy controls [Kalla et al. 2007]. Their results also confirmed that 4-AP decreased DBN during gaze straight ahead in 12 of the 15 patients, and especially reduced spontaneous upward drift in four patients with degenerative CAs. They concluded that 4-AP improved fixation by restoring gaze-holding ability.
Mechanisms of action
Restoration of flocculus/paraflocculus function
Vestibulocerebellar lesions are thought to be the main etiology of DBN [Pierrot-Deseilligny and Milea, 2005; Kalla et al. 2007]. Previous studies in monkeys showed that bilateral ablation of the flocculus and paraflocculus elicited DBN [Zee et al. 1981]. Furthermore, in a patient with DBN and CAs, Bensea and colleagues demonstrated reduced cerebral glucose metabolism bilaterally in the region of the cerebellar tonsil and flocculus/paraflocculus and that treatment with 15 mg/day of 4-AP lessened hypometabolism and simultaneously improved DBN [Bensea et al. 2006]. Taken together, floccular and parafloccular dysfunction seems to be responsible for the development of DBN; 4-AP successfully restores ocular coordination in these areas.
No clear mechanism for DBN improvement at neural circuitry level
It has been assumed that reinforcement of PC excitability in the flocculus/paraflocculus may improve DBN [Kalla et al. 2007]. However, taking into consideration the finding that 4-AP at therapeutic concentrations fails to increase output [Alvina and Khodakhah, 2010], it is likely that the mechanism of action of 4-AP is more complex. While the neural mechanism underlying DBN remains obscure, several explanations have been proposed for the spontaneous upward drift: tonic imbalance of the central vestibular pathway in vertical eye movements [Baloh and Spooner, 1981]; imbalance of the vertical smooth pursuit tone [Zee et al. 1981]; defective function of the neural velocity-to-position integrator for gaze holding [Glasauer et al. 2003]; and on-directions asymmetry (only ~10% of PCs have on directions for upward gaze velocity) [Marti et al. 2005]. Still, further physiological in vitro and in vivo studies are necessary to identify the exact cerebellar dysfunctions that elicit DBN, and the mechanisms of action of aminopyridines associated with restoration of these changes.
Degenerative cerebellar ataxias
Efficacy of aminopyridines in this group of CAs remains controversial. Randomized, double-blind studies are lacking. The efficacy of 4-AP at 10 mg twice daily was examined in 16 patients with SCA1, 3, and 6 [Giordano et al. 2013]. Significant improvement was observed in the 8 m walking test and speech rate test, but not in the SARA score and nine-hole peg test. The authors concluded that 4-AP can induce short-term (14 days) improvement in CAs. In contrast, others reported that, while treatment with 3, 4-DAP at 20 mg twice daily improved DBN, it had no beneficial effects on other CAs in 10 patients with SCA6 and 5 patients with 16q22.1-linked ADCA [Tsunemi et al. 2010]. The same treatment also had no effect on international cooperative ataxia rating scale (ICARS) and postural sway. The contradictory results might be due to differences in the effects of 4-AP on each clinical cerebellar sign. In this regard, Schniepp and colleagues reported that 4-AP at 5 mg three times daily improved gait variability in stepping in 31 patients with various pathologies, without improvements in gait speed or cadence, both of which reflect stability [Schniepp et al. 2012]. In agreement with the observed variability in the efficacy of 4-AP on different clinical cerebellar signs, it is noteworthy that stepping and stability are controlled separately and independently in the cerebellum [Ienaga et al. 2006]. Further randomized, double-blind studies are necessary to determine the effects of aminopyridines on specific aspects of CAs.
Neuromodulation therapy using other drugs
Clinical evidence
Several studies have reported the efficacies of other drugs on degenerative CAs (Table 3). For example, a few randomized double-blind and placebo-controlled studies have shown significant improvements in clinical ataxic scales after treatment with riluzole, an agonist of small conductance potassium channel and an inhibitor of glutamate release, and varenicline, an agonist of α4β2 nicotinic acetylcholine receptors [Ristori et al. 2010; Romano et al. 2015; Zesiewicz et al. 2012]. Furthermore, two open-label studies showed that acetyl-DL-leucine and acetazolamide improved SARA score and Ataxic Rating Scale, respectively [Strupp et al. 2013; Yabe et al. 2001]. However, the extent of score change was not large enough to improve the daily lives of patients. Thus, the efficacies of these drugs in CAs, especially in terms of improvements in activities of daily life, need to be confirmed in large randomized double-blind and placebo-controlled studies.
Drug trials in patients with degenerative cerebellar ataxias.

ICARS, International Cooperative Ataxia Rating Scale; SARA, Scale of Assessment and Rating of Ataxia.
Mechanisms of action
Mechanisms of actions are not fully understood. The efficacies of these drugs seem to be mediated through multiple mechanisms [Ilg et al. 2014; Romano et al. 2015]. Of these diverse and heterogeneous actions, the following two assumptions form the basis of a plausible working hypothesis: adjustment of neuronal activities in DCN; and augmentation of cerebellar functions through modulatory afferent systems (cholinergic afferents).
It is assumed that riluzole restores an exaggerated firing rate of CN neurons [Ilg et al. 2014]. Since CN neurons receive profound inhibitory outputs from the cerebellar cortex via PCs, degeneration of PCs could elicit hyperactivities in CN neurons (see Figure 3). However, recent physiological studies have described a critical role for the neuronal activity in the dentate nucleus (DN, one of DCN) in coordinating the control of movements of the upper limbs [Ishikawa et al. 2014]. For example, the release of DNs generated by reduced inhibition from PCs, that is disinhibition, facilitates the execution of wrist movements, while suppression of the DNs by increased PC activity contributes to the stabilization of proximal muscles and improves task performance (see Figure 3). Thus, exaggerated activities of CN neurons might interfere with the production of appropriate cerebellar outputs using disinhibition or inhibition circuit mechanism, leading to limb CA [Ishikawa et al. 2014]. Under these conditions, adjustment of DCN neuronal activity could be a therapeutic target (see Figure 3). Similarly, acetyl-DL-leucine modulates the activities of ventral vestibular neurons [Vibert and Vidal, 2001], which might contribute to adjustment of neural activity in the vestibular cerebellar cortex.
Varenicline might also facilitate cerebellar functions. Although the cerebellum receives relatively few cholinergic projections, fibers originating from the vestibular nuclei terminate in the nodulus and ventral uvula and use acetylcholine as neurotransmitter or coneurotransmitter [Jaarsma et al. 1997]. Application of carbachol, a nicotinic and muscarinic receptors agonist, in the rabbit flocculonodular lobe increased the amplitude of the vestibulo-ocular reflex [Tan and Collewjin, 1991]. These physiological findings coincide with the clinical finding of varenicline-induced improvement in gait scores [Zesiewicz et al. 2012].
Summary for anti-cerebellar ataxia medications
The results of experimental and clinical studies on EA2 suggest that distortion of PC pacemaking is a potential therapeutic target in CAs. Aminopyridines restore the pacemaking and result in clinical improvement of CAs in patients with EA2. However, the efficacy of aminopyridines in CAs remains controversial, with the exception of the clinical signs of DBN and stepping irregularities in gaits. Several placebo-controlled clinical trials are currently underway to determine the therapeutic effects of a sustained form of 4-AP in patients with EA2 and patients with ataxic gaits [Ilg et al. 2014].
In EA2, mutations in the P/Q-type voltage-gated Ca2+ channel elicit KCa channel dysfunction, leading to distortion of PC pacemaking. Thus, aminopyridines restore specific etiology itself (P/Q-type-voltage-gated Ca2+–KCa channels–PC pacemaking system) in EA2. In contrast, in degenerative CAs, aminopyridines are expected to reinforce a number of lost cerebellar functions mainly by potentiation of one cerebellar element, PC pacemaking, alone. Interestingly, nystagmus and gait stepping, which improve following treatment with aminopyridines, share common rhythm-related properties. Taken together, the difference in therapeutic roles might explain the difference in efficacies of aminopyridines in EA2 and degenerative CAs.
So far, excepted for aminopyridines, there is no convincing evidence for any therapeutic effects at activities of daily life level for other drugs. However, few clinical data suggest potential efficacies in adjustment of CN neuronal activities (an element underlying cerebellar outputs) and activation of cholinergic facilitative afferents. Further studies should be conducted.
Noninvasive cerebellar stimulation
Clinical evidence
The anatomical location of the cerebellum below the skull is particularly well suited for noninvasive electrical stimulation and, therefore, it is not surprising that several trials are ongoing in various forms of CAs.
Benussi and colleagues have studied the effects of a single session of anodal transcranial direct current stimulation (anodal tDCS) in 19 patients with ataxia in a double-blind, randomized, sham-controlled study [Benussi et al. 2015]. The authors studied clinical scores (SARA, ICARS) as well as the nine-hole peg test, and the 8 m walking time. The authors found a positive effect in terms of performances compared with the sham trial.
In two patients presenting with SCA2, the combination of tDCS of the cerebellum and the motor cortex (tCCDCS: transcranial cerebello-cerebral DC stimulation) was associated with a reduction of postural or action tremor as well as hypermetria [Grimaldi et al. 2014b]. An effect on the timing of antagonist muscle commands was observed.
Despite these positive effects on ataxia, we are still lacking rigorous large-scale clinical trials. Given the heterogeneity of CAs, this is mandatory.
Mechanisms of action
There is a consensus that both transcranial magnetic stimulation (TMS) and tDCS can effectively influence cerebellar functions in the motor and cognitive domains [Grimaldi et al. 2014a]. In the motor domain, effects on motor adaptation and learning, visually guided tracking tasks, and motor surround inhibition have been observed. For the cognitive operations, effects on verbal working memory, semantic associations and predictive language processing have been observed. Both TMS and tDCS tune the excitability of the cerebellar cortex, which impacts on the degree of inhibition of cerebellar nuclei. Applying TMS over the cerebellum induces changes in the amplitudes of motor evoked potentials elicited from the primary motor cortex (M1) as demonstrated by Ugawa in the so-called paradigm of cerebellum-brain inhibition (CBI) [Ugawa et al. 1995]. tDCS induces a polarity-dependent site-specific modulation of cerebellar activity not only from the electrical standpoint but also by impacting on the metabolism [Grimaldi et al. 2016]. Cathodal tDCS over the cerebellum decreases the ability of TMS to elicit CBI of M1, whereas anodal tDCS exerts the opposite effect [Galea et al. 2009].
The modulation of CBI by tDCS is not accompanied by changes in spinal or brainstem excitability. The possibility to tune noninvasively the activity of the cerebellar cortex, in particular the Purkinje neurons which control the nucleo-thalamic drive, and the observation of physiological effects on locomotor adaptation [Jayaram et al. 2012] open strong perspectives for the management of gait ataxia, which is one of the most disabling symptoms in cerebellar patients.
Repeated TMS can also affect CBI. For instance, continuous theta burst stimulation can suppress CBI [Carrillo et al. 2013]. However, clinically relevant studies in terms of efficacy against ataxic deficits are lacking.
Overall, it is concluded that tDCS has a potential therapeutic role in the symptomatic management of motor or cognitive deficits in patients with CAs, but several key questions remain unanswered. What is the optimal current and where exactly should it be applied over the cerebellum? How many stimulation sessions should be delivered? Which subsets of patients are responsive?
Motor rehabilitation
Clinical evidence
Several lines of evidence indicate the efficacy of motor rehabilitation in CAs elicited by limited lesions in the cerebellum (e.g. vascular diseases, tumors, MS). However, whether motor rehabilitation can improve symptoms of degenerative CAs remains a matter of debate. This is especially true when the insult is rapidly progressive in nature and affects the entire cerebellum.
Two recent cohort studies have provided evidence for the effectiveness of motor rehabilitation in degenerative CAs [Ilg et al. 2009, 2010; Miyai et al. 2012]. These studies were conducted in 16 patients with ataxia (age: 61.4 ± 11.2 years; 10 patients with degenerative CAs and 6 patients with sensory ataxia; disease duration: 12.9 ± 7.8 years; baseline SARA score: 15.8 ± 4.3) [Ilg et al. 2010] and 42 patients with ataxia (age: 62.5 ± 8.0 years; all had degenerative CAs; disease duration: 11.3±3.8 years; baseline SARA score: 11.3 ± 3.8) [Miyai et al. 2012]. The results showed that motor rehabilitation produces the following results:
Four-week intensive rehabilitation (1 h × 3 per week; 2 h × 5 + 1 h × 2 per week) [[Ilg et al. 2010; Miyai et al. 2012] improved balance, gait and interlimb coordination in SARA score, and in score for activities of daily living by a Functional Independence Measure. Improvement in trunk ataxia in the SARA score was more impressive than in limb ataxia.
Four-week intensive rehabilitation is more effective in patients with mild atrophy than in patients with severe atrophy.
Persistent improvement was noted up to 12 weeks, but not 24 weeks, after intensive rehabilitation, following cessation of rehabilitation.
Long-term program of 1 h homework training per day was associated with 1-year benefits, despite gradual decline in motor performance associated with natural progression of the disease.
Low-intensity homework training up to 30 min per week failed to maintain improvement.
Even 26 weeks after the first intensive rehabilitation, reboost of rehabilitation induced similar improvement compared with the first intensive rehabilitation.
Thus, these results show the efficacy of intensive rehabilitation therapy, especially when combined with reboost rehabilitation in degenerative CAs.
Mechanisms of action
Motor rehabilitation reinforces cerebellar motor learning and development of a new internal model
Recent physiological studies have provided convincing evidence that the cerebellum coordinates movements in a predictable fashion [Manto et al. 2012]. According to the internal model theory [Kawato et al. 1987], neural mechanisms that mimic the input or output characteristics of a motor apparatus are assumed to be embedded in the cerebellum. The forward internal model can predict sensory consequences in response to issued motor commands, whereas the inverse internal model can calculate the predicted motor commands from desired movements. The internal model is acquired through motor learning, which leads to acquisition of skillful movements [Deuschl et al. 1996]. Various types of cerebellar synaptic circuitries contribute to motor learning. For example, the Marr–Albus–Ito hypothesis proposed that error signals from climbing fibers decrease parallel fiber inputs onto PCs [Ito, 2006] and thus eliminate inadequate outputs (see Figure 3). However, this hypothesis remains to be approved [Manto, 2008]. Although these theories of internal model and motor learning have not yet been confirmed with certainty in real cerebellar neural circuits [Manto et al. 2012], the efficacy of motor rehabilitation can be explained by these hypotheses. Motor rehabilitation seems to promote motor learning and establishing a new internal model in the residual and intact portion of the cerebellum, leading to compensation of the impaired coordinated movements.
Motor rehabilitation facilitates change in strategies for motor control
An alternative mechanism for the efficacy of rehabilitation is a change in strategy for motor control. Motor rehabilitation includes a combination of restorative and compensatory techniques [Ilg et al. 2014]. For acquisition of compensatory techniques, patients are trained to replace rapid multi joint movements with sequential single joint movements, and to walk under careful visual-guided movements, instead of predictive walking [Ilg et al. 2014]. In these compensatory movements, motor commands are calculated without cerebellar involvements. Patients will try to perform movements under a new strategy that does not involve the cerebellum.
In this regard, intention tremor or action myoclonus, a cerebellar sign induced by cerebellar efferent systems, occurs in visually guided movements, in which the cerebellum operates according to predictive controls for exact trace. However, memory-guided movements are diminished due to lesser involvement of the cerebellum in such movements [Mitoma et al. 1993].
Taken together, using compensatory techniques (change in motor strategy), the physical activities of daily living could improve without improvement in cerebellar functions.
Conclusion
Early therapy during ‘restorable stage’ of preserved cerebellar functions
Various treatments directed towards the cause of CA have been designed for patients with malformations, vascular diseases, neoplasms, metabolic diseases, and IMCAs. The aim of such therapies is to stop disease progression (minimize cell loss and preserve cerebellar function). The preservation of cerebellar function is physiologically identified in the early stage of IMCAs [Mitoma et al. 2016]. Using a technique based on the ratio of feedforward or feedback control, we quantified and compared the control modes in patients with early IMCAs and those with degenerative CAs [Mitoma et al. 2016]. Patients with early IMCAs showed no or slight atrophy, suggesting minimal cell death. The operation of feedforward control could be an index of survival of cerebellar function. Interestingly, motor control in patients with early IMCAs was opposite to that in patients with degenerative CAs, although both groups of patients showed similar clumsiness in tracking tasks. The cerebellar feedforward control is still present in the former group, though with ill-defined property, whereas it is abolished in the latter (Figure 4).

A scheme of the decline of cerebellar functions and the concept of ‘restorable stage’. Proper therapies could restore cerebellar functions in patients whose cerebellum is at ‘restorable stage’, meaning that there is still a sufficient preservation of cerebellar functions. After a given threshold of neuronal loss or dysfunction in the cerebellar circuitry, cerebellar functions cannot be restored anymore because the loss of computational capacities of the remaining cerebellar modules is too severe. Motor Cx, motor cortex.
Consistent with these physiological data, the control of autoimmunity by immunotherapy is followed by two types of clinical courses in IMCAs [Mitoma et al. 2015]: patients with early stage CA (without cerebellar atrophy) showed full or partial recovery, whereas patients with advanced stage CA (with moderate cerebellar atrophy) showed persistent CAs, though without progression. Similar responses are also observed in neuromodulation therapies. Motor rehabilitation is more effective in patients with mild atrophy than in patients with severe atrophy [Ilg et al. 2010; Miyai et al. 2012]. Taken together, the difference in the clinical course is probably due to differences in compensatory abilities in the residual cerebellum.
In CAs associated with other diseases, proper therapies could also restore cerebellar functions in patients at ‘restorable stage’ with sufficient preserved cerebellar functions. Thus, early diagnosis and therapy are important to prevent worsening beyond the ‘restorable stage’. In this physiological scheme, there is a minimal threshold for the predictive controller (a dotted line in Figure 4). From the clinical standpoint, the rationale for a threshold is for instance that patients with slowly progressive tumors may be remarkably asymptomatic for a long time before manifesting CA [Dow and Moruzzi, 1958; Manto, 2002]. The minimal threshold would reflect ‘cerebellar reserve’. Thus, techniques such as transplantation of stem cells aim to increase the minimal threshold (in Figure 4, a downward shift of a dotted line) by reconstruction of entire cerebellar circuits and reinforcement of ‘cerebellar reserve’.
Understanding the efficacy of neuromodulation therapy based on cerebellar function
Many assumptions have been proposed to clarify the role of the cerebellum in the coordination of movements [Manto et al. 2012]. Two main models have been proposed to explain the principle of the cerebellar neural machinery [Galliano and De Zeeuw, 2014]. The hypothesis of Braintenberg considers the cerebellum as a timing control machine [Braitenberg and Atwood, 1958]. This model assumes that the climbing fiber mediated inputs synchronize the activity of PCs for fine tuning of timing (Figure 3) [Llinas, 2009]. In contrast, the Marr–Albus–Ito hypothesis considers the cerebellum as a learning machine [Ito, 2006]. This model proposes that the climbing fiber mediated inputs, which convey error signals, depress inappropriate parallel fiber mediated inputs through long-term depression, so as to formulate an internal model (Figure 3) [Ito, 2006]. There is no integral theory that covers these two divergent hypotheses. However, the efficacy of aminopyridines could be explained by the timing control machine model, whereas the efficacy of motor rehabilitation could be explained by the learning machine model.
However, irrespective of the operation principle, the on/off signals are formulated in CN neurons (these cells are the output neurons of the cerebellum) through the mechanism of disinhibition or inhibition on CN neurons from the cerebellar cortex neurons (Figure 3) [Ishikawa et al. 2014]. Importantly, two separate groups of neurons, the cerebellar cortex neurons (granule cells, PCs, and inhibitory interneurons) and CN neurons, formulate cooperatively on/off cerebellar output signals. This geometrical structure, crystal-like architecture [Galliano and De Zeeuw, 2014] allows clinicians to potentiate impaired on/off cerebellar outputs by facilitating the cerebellar cortex with noninvasive stimulation. Some drugs, riluzole and acethyl-DL-leucine, which adjust the activities of CN neurons, might also restore the capacity of the generation of the on/off signal in the cerebellum.
In conclusion, the understanding of the physiological basis of the various therapies is a critical step for clinicians dealing with CAs. We suggest that some degree of reversibility can be achieved if the therapies of CAs are administered as early as possible and take into account the pathogenesis behind the disorder. Novel therapies should take into account the mechanisms of the cerebellar circuitry in order to be effective. The trial and error strategy that has prevailed in CAs should be abandoned.
Footnotes
Funding
MM is supported by the FNRS-Belgium.
Conflict of interest statement
The authors declare that they have no conflict of interest.