
Abstract
In this paper, we review the experience with fenfluramine in epileptic and other paroxysmal disorders. Since the best available data are from the treatment of Dravet syndrome, we will focus primarily on this condition. Originally fenfluramine was launched as an anorectic agent. As early as 1985, seizure reduction in children could be demonstrated in a few cases with photosensitive, self-induced epilepsy. Hereafter, a small study was launched in patients with self-induced epilepsy. Results showed a significant seizure reduction, and review of the patient data showed that 5 of the 12 patients had Dravet syndrome. During that observation period, fenfluramine was withdrawn from the market because of cardiovascular side effects associated with prescribing higher doses in combination with phentermine for weight loss. In March 2002, a Belgian Royal Decree was issued permitting further study of fenfluramine in pediatric patients with intractable epilepsy. In 2011 under the Royal Decree, a prospective study of patients with Dravet syndrome treated with low-dose fenfluramine was initiated and is currently ongoing. The initial results are promising in terms of reduction of seizure frequency and overall tolerability.
Dravet syndrome
Charlotte Dravet first described the core clinical features of a rare and severe epileptic syndrome in 1978 [Dravet, 2011]. Dravet syndrome, previously known as severe myoclonic epilepsy of infancy, is considered an encephalopathy because the epileptic activity itself is believed to contribute to severe cognitive and behavioral impairment above and beyond what might be expected from the underlying pathology [Berg et al. 2010]. Children with Dravet syndrome are typically healthy and developmentally normal infants who present in infancy with recurrent seizures, most commonly provoked by fever. As compared with non-Dravet syndrome patients, these seizures tend to have an earlier presentation (before 7 months) and longer duration (>10 minutes), occur more frequently (often ⩾5 in infancy), and consist of hemiconvulsions, myoclonic seizures, or focal seizures [Hattori et al. 2008]. The interictal electroencephalography (EEG) and central imaging in general are normal during the first year. Within the second year of life, a developmental arrest (or regression) becomes evident, and during the following years multiple other therapy-resistant seizure types appear. Over time, the interictal EEG can remain normal or show nonspecific features such as background and epileptiform discharges [Specchio et al. 2012; Lee et al. 2015].
The diagnostic criteria for Dravet syndrome are based on the clinical phenotype and include the child’s age of seizure onset, evolution of seizure types, EEG features, and developmental course [Dravet, 2011; Scheffer, 2012]. More recently, genetic evidence supportive of diagnosis was present in approximately 75% of the patients, with the most frequent mutations occurring in the SCN1A gene. The SCN1A gene codes for the α1 subunit of the voltage-gated sodium channel Nav1.1, which is required for the generation and propagation of action potentials throughout the central nervous system (CNS) [Bender et al. 2012]. Data from SCN1A knockout mice showed that the α subunit is fundamental for the excitability of hippocampal GABAergic interneurons [Mistry et al. 2014]. Reduced sodium currents in these inhibitory interneurons enhance the excitability of their downstream synaptic targets (i.e. pyramidal neurons), which may lead to epilepsy [Yu et al. 2006; Ogiwara et al. 2007; Rubinstein et al. 2015].
The majority of patients with an SCN1A mutation have a truncating or missense mutation. In 3–5% of patients with Dravet syndrome, a copy number variant (CNV), most frequently involving deletions, is found [Marini et al. 2009, 2011; Suls et al. 2013]. Mutations in SCN1A are not only associated with Dravet syndrome but with a variety of other epilepsies, familial hemiplegic migraine, and autism [Weiss et al. 2003; Cestele et al. 2008]. Meng and colleagues have investigated 1257 mutations of the SCN1A gene and their relationship with functional alteration of SCN1A and the subject phenotype [Meng et al. 2015]. As demonstrated by previous studies [Ceulemans et al. 2004; Mulley et al. 2005], a more severe phenotype is associated with severe functional alteration of the Nav1.1 channel. Patients with Dravet syndrome frequently had a de novo mutation which lead to a loss of function of the Nav1.1 channel; for example, a missense mutation of the pore region. Other genes have also been reported as involved in the spectrum of Dravet syndrome, including GABRG2 [Harkin et al. 2002], PCDH19 [Depienne et al. 2009], SCN1B [Patino et al. 2009], and CHD2 [Suls et al. 2013]. Nevertheless about 25% of patients with Dravet syndrome remain without an identified genetic mutation.
The discovery of SCN1A mutations as the primary genetic cause of Dravet syndrome has led to a better understanding of its etiology and treatment [Claes et al. 2001]. The treatment strategy is focused on three main principles. (1) Prevention of febrile seizures by preventing hyperthermia. Since body temperatures above 37°C can trigger convulsions in Dravet patients, hot baths, excessive ambient warmth or physical exercise on sunny days have to be avoided. Logically fever has to be adequately treated with antipyretics [Verbeek et al. 2015]. (2) Use of adequate rescue treatment with benzodiazepines. To prevent long-lasting status epilepticus parents and caregivers have to be trained to administer benzodiazepines as soon as possible. All Dravet patients should have an emergency protocol to guide the emergency doctor in the treatment of a status epilepticus. Phenytoin, phenobarbital and thiopental should be avoided as they can worsen the outcome. (3) Rational and adequate maintenance therapy with antiepileptic drugs (AEDs) with avoidance of specific sodium channel-blocking agents, such as lamotrigine, carbamazepine, and phenytoin, as they are known to aggravate the symptoms [Guerrini et al. 1998; Genton, 2000]. At present, the standard treatment in Europe is either the combination of stiripentol (STP), valproate (VPA) and clobazam (CLB), or VPA in combination with topiramate (TPM) [Chiron and Dulac, 2011]. STP, which is structurally unrelated to any other AED, is currently the only registered drug in Europe for the treatment of Dravet syndrome and has had orphan drug status for this indication in Europe since 2001; it has not been approved by the United States Food and Drug Administration (FDA). STP is believed to act by two main mechanisms: (1) direct GABAergic effect [Quilichini et al. 2006] and (2) inhibition of the cytochrome P450 system, which indirectly increases the concentration of many AEDs, including CLB [Giraud et al. 2006]. Placebo-controlled trials of STP in Dravet syndrome showed that STP (in combination with VPA and CLB) is significantly better than placebo with regards to a ⩾50% reduction in seizure frequency and seizure freedom [Brigo and Storti, 2013]. TPM has been shown to be effective in two open-label studies [Nieto-Barrera et al. 2000; Coppola et al. 2002] and in patients with poor control on STP [Kroll-Seger et al. 2006]. Other smaller studies reported an effect of levetiracetam [Striano et al. 2007], ketogenic diet [Dressler et al. 2015], and vagal nerve stimulation [Zamponi et al. 2011]. Despite the better approach and earlier recognition, the outcome in the vast majority of Dravet patients is still poor with multiple drug-resistant seizures, moderate to severe intellectual disability, behavioral issues, and motor abnormalities [Takayama et al. 2014]. As epileptic seizures have a negative impact on the quality of life [Brunklaus et al. 2011] and probably on cognitive development [Wolff et al. 2006], other therapeutic treatments are needed.
Fenfluramine
Fenfluramine (3-trifluoromethyl-N-ethylamphetamine), a derivate of amphetamine, was recognized as an appetite suppressant in the mid-1960s [Munro et al. 1966] and approved for this indication in the US in 1973. As a derivative of amphetamine, the anorectic effect was believed to be exercised through serotoninergic mechanisms. Indeed, fenfluramine causes the release of serotonin (5-HT) by disrupting vesicular storage of the neurotransmitter and inhibiting its reuptake [Fuller et al. 1988]. The increased 5-HT availability in the brain results in a loss of appetite. By the 1980s, fenfluramine was being used by more than 60 million people worldwide. It was frequently combined ‘off-label’ with monoamine oxidase inhibitor phentermine to sustain the effect of fenfluramine, lending to the famous ‘Fen–Phen’ combination.
Cardiovascular safety concerns associated with fenfluramine emerged in the 1980s when the first two cases of a possible relationship between the use of fenfluramine and pulmonary hypertension (PH) were reported [Douglas et al. 1981]. In 1996, a rigorous case–control study comparing 95 patients with definite or probable PH with 355 control patients matched to individual case patients by age, sex, and number of annual physician visits found that the use of appetite-suppressant drugs (most commonly dexfenfluramine and fenfluramine) for ⩾3 months was associated with an odds ratio for PH of 23 (95% confidence interval [CI] 6.9–77.7) [Abenhaim et al. 1996]. Valvular heart disease in patients treated with the ‘Fen–Phen’ combination was first described by Connolly and colleagues [Connolly et al. 1997]. The US FDA announced the withdrawal of fenfluramine and dexfenfluramine shortly thereafter due to this increased risk. However, because fenfluramine exhibits a stimulation of the central serotoninergic pathways, other therapeutic indications continued to be investigated, including epilepsy.
Serotonin and its working mechanism in epilepsy
The functions of 5-HT on the CNS are numerous and appear to involve control of appetite, sleep, cognitive functions, mood, (social) behavior, cardiovascular function, muscle contraction, endocrine regulation, maturation of neuronal and glial cells, synaptic connections, and epilepsy [Filip and Bader, 2009]. Bonnycastle and colleagues were the first to suggest the link between elevations in CNS 5-HT levels and inhibition of epilepsy [Bonnycastle et al. 1957]. In their study, they demonstrated that several anticonvulsant agents elevated brain 5-HT concentrations in rats [Bonnycastle et al. 1957]. In general, agents that elevate extracellular 5-HT levels, such as 5-hydroxytryptophan and 5-HT reuptake blockers, inhibit both focal and generalized seizures [Jobe and Browning, 2005], as shown in different animal models [Dailey et al. 1992; Yan et al. 1994; Pasini et al. 1996]. Conversely, depletion of brain 5-HT seems to lower the threshold to audiogenically, chemically, and electrically evoked convulsions [Bagdy et al. 2007; Trindade-Filho et al. 2008; Da Fonseca et al. 2015]. Several different 5-HT receptors are expressed on cortical and/or hippocampal glutamatergic or GABAergic neurons or terminals [Barnes and Sharp, 1999]. These receptors can directly or indirectly change the ion conductance or concentration within cells, resulting in de- or hyperpolarization of neurons, and thus influence the excitability of the neural networks involving epilepsy. Although 5-HT proved to have an antiepileptic effect in animal models [Hamid and Kanner, 2013], no randomized clinical trials of agents with serotoninergic mechanisms have been reported.
Additional support for the role of 5-HT in epilepsy is provided by clinical studies of selective serotonin reuptake inhibitors (SSRIs) in humans, which showed a possible antiepileptic effect [Favale et al. 1995, 2003; Specchio et al. 2004]. Review of the literature revealed three open-label clinical studies with SSRIs in subjects with epilepsy [Favale et al. 1995, 2003; Specchio et al. 2004]. Specchio and colleagues [Specchio et al. 2004] studied the safety of citalopram as add-on therapy in patients with relatively well-controlled seizures and comorbid depression. Although the study primarily was designed to demonstrate safety, the patients with citalopram showed a statistically significant improvement of seizures independent of the seizure type. Favale and colleagues examined the efficacy of citalopram in 11 nondepressed patients with poorly controlled focal epilepsy [Favale et al. 2003]. Nine of the 11 patients demonstrated a reduction in seizure frequency of ⩾50%. Favale and colleagues also conducted an open-label trial with fluoxetine in 17 patients with focal epilepsy (with or without secondary generalization) [Favale et al. 1995]. Six patients became seizure-free during the mean follow-up of 14 months and the remaining 11 patients had an average 30% reduction in seizure frequency. In 2014, Meador [Meador, 2014] published the case of an adult woman with Dravet syndrome and invalidating stereotypic behaviors. A trial with fluoxetine led to a marked reduction of the stereotypic behaviors and a clinically significant reduction in frequency and severity of seizures.
Fenfluramine in epilepsy and other paroxysmal disorders
Experiments
Through its central serotoninergic activity fenfluramine was suspected to have an anticonvulsive effect; however, the exact mechanism of action is yet to be fully characterized. Further research is required to determine whether other known anticonvulsant receptors are also involved. Gentsch and colleagues [Gentsch et al. 2000] reported that fenfluramine blocked epileptiform activity in perfused rat entorhinal cortex brain slices induced by lowering the magnesium concentration in the artificial cerebrospinal fluid. Application of a 5-HT1A receptor antagonist diminished the effect of fenfluramine, as did paroxetine, a 5-HT reuptake inhibitor. Fenfluramine releases 5-HT through a carrier-dependent mechanism which can be inhibited by prior treatment with a 5-HT reuptake blocker [Fuller et al. 1988]; this inhibition may represent competition between substrates for the (re)uptake carrier. Zhang and colleagues recently demonstrated the anticonvulsive effect of fenfluramine in an antisense knockdown zebrafish model of Dravet syndrome. Incubation with fenfluramine significantly reduced the hyperactivity and the epileptiform discharges of the scn1Lab morphants [Zhang et al. 2015].
Early case reports and trials
The published data describing the use of fenfluramine in neurological disorders are limited to case reports and several small open-label studies; these are summarized in Table 1. Gastaut described the use of fenfluramine in three children with self-induced syncopes [Gastaut, 1984]. The addition of fenfluramine (30–60 mg/day) led to a disappearance of the syncopes and an amelioration of their autistic behavior. Aicardi and Gastaut described the use of fenfluramine (60 mg/day) in three adolescent patients with intractable self-induced photosensitive epilepsy [Aicardi and Gastaut, 1985]. All three patients demonstrated a significant reduction in seizure frequency; in one patient, a 20-year old male, the EEG no longer showed evidence of photosensitivity. The investigators proposed a dual mechanism for fenfluramine: (1) antipsychotic mechanism that suppressed the compulsive need for self-induction of seizures; and (2) direct antiepileptic mechanism. Aicardi and colleagues reported [Aicardi et al. 1988] the case of an 11-year old girl with self-induced syncopal episodes caused by a Valsalva maneuver. In addition, the girl had typical absences that occurred spontaneously or were triggered by hyperventilation or self-induced apneas. Treatment with VPA suppressed the absence seizures but had no effect on the compulsive respiratory stereotypies. The addition of fenfluramine (60 mg daily) led to a disappearance of these self-induced apneas along with a marked improvement of the behavior, and discontinuation of the treatment was associated with a return of the attacks.
Summary of studies of fenfluramine in epilepsy.
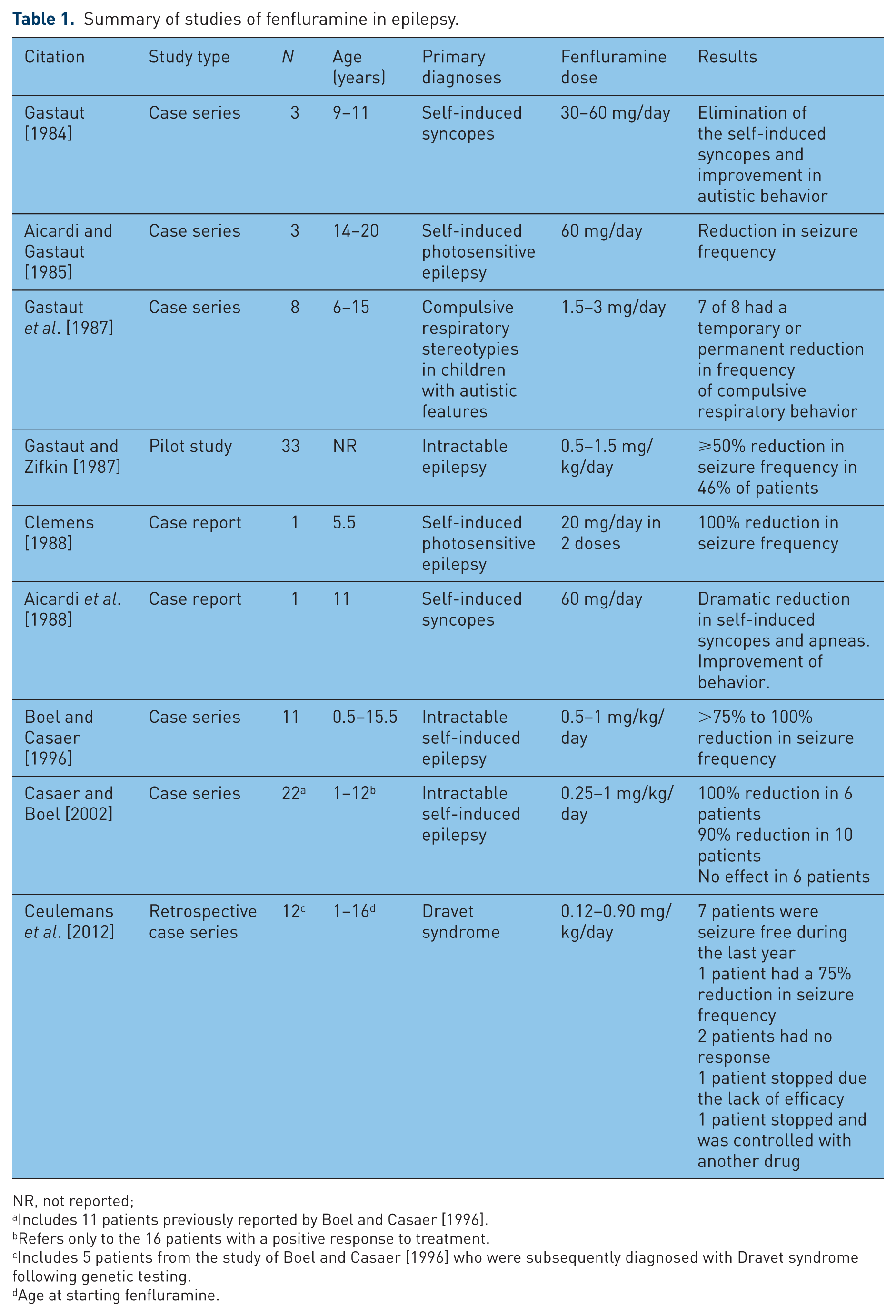
NR, not reported;
Includes 11 patients previously reported by Boel and Casaer [1996].
Refers only to the 16 patients with a positive response to treatment.
Includes 5 patients from the study of Boel and Casaer [1996] who were subsequently diagnosed with Dravet syndrome following genetic testing.
Age at starting fenfluramine.
Gastaut and colleagues reported the results of an open trial of fenfluramine in eight patients with autism and syncope due to compulsive respiratory stereotypies [Gastaut et al. 1987]. Fenfluramine was administered at doses between 1.5 and 3 mg/kg/day. For four of the five patients with frequent Valsalva maneuvers, the respiratory stereotypies and syncopes were suppressed for 2–18 months. One girl with Lennox–Gastaut syndrome demonstrated a two-thirds reduction in seizure frequency during treatment. In addition, one of the two patients with generalized spike-wave complexes in the EEG showed a significant amelioration. Adverse events included sleepiness, which was seen in two patients and resolved after lowering of the dose, and early weight loss which was seen in two patients and stabilized after a loss of 2 kg. Further support for the antiepileptic activity of fenfluramine was provided by a several small open-label studies. In 33 patients with severe childhood epilepsy [Gastaut and Zifkin, 1987], fenfluramine was added to the patient’s current antiepilepsy treatment regimen and nearly half of the patients (46%) responded with a >50% reduction in seizure frequency. Boel and Casaer [Boel and Casaer, 1996] treated 11 children (ages 18 months to 15.5 years old) with refractory or self-induced epilepsy with fenfluramine at 0.5–1 mg/kg/day for 3–8.5 years. Seven children (64%) became seizure-free and the other four experienced ⩾75% reduction in seizure frequency. All patients had a transient loss of appetite but without weight loss. Slight sedation occurred in six children but was managed by dividing the daily dose into two or three individual doses. No cardiologic examinations were performed. In 2002, Casaer and Boel [Casaer and Boel, 2002] published a brief update of their study with fenfluramine. The study population was expanded to 22 patients, including the previously reported 11 patients. All patients had self-induced seizures. Six patients (27%) had a complete seizure control (seizure-free period ranging from 1 to 12 years). Ten patients (45%) had a 90% seizure reduction and six patients (27%) showed no improvement and discontinued therapy [Casaer and Boel, 2002]. The side effects were limited to transient loss of appetite and fatigue after a physical effort, both reported in one patient. No cardiologic examinations were performed.
In 1988 Clemens reported the case of a boy of 5.5 years of age with Lennox–Gastaut syndrome and self-induced seizures resistant to conventional anticonvulsive treatment. The addition of bromocriptine led to a seizure reduction but the combination with fenfluramine was able to terminate the self-induced seizures by means of blocking the photosensitive triggering mechanism [Clemens, 1988].
Fenfluramine in Dravet syndrome
Retrospective data
Ceulemans and colleagues reported on the effect of fenfluramine in 12 patients with Dravet syndrome [Ceulemans et al. 2012], retrospectively analyzed in 2010. The patient population included five patients from a study reported by Boel and Casaer [Boel and Casaer, 1996] who after review of their files and genetic testing were diagnosed with Dravet syndrome. Eleven of the patients had a mutation in SCN1A. In 2001, during the observation period of this retrospective study, fenfluramine was withdrawn from the market and a subsequent Belgium Royal Decree was issued in March 2002 allowing the use of fenfluramine in clinical studies of refractory epileptic seizures.
All patients in the study had drug-resistant epilepsy with multiple seizure types. Fenfluramine treatment was started at a mean age of 8 years (range: 1–16 years), administered at a mean of 0.34 mg/kg/day (range: 0.12–0.90), and added to each patient’s current antiepilepsy regimen. The mean follow-up time after starting fenfluramine was 11 years and 4 months (range: 1–22 years).
At the time of the present analysis, fenfluramine had been stopped in two patients. One patient stopped treatment due to lack of efficacy, and the other discontinued fenfluramine after its withdrawal from the market and remained seizure-free with VPA monotherapy. Seven of the remaining 10 patients (70%) had been seizure-free for at least 1 year prior to their most recent study visit, with a mean seizure-free period of 6.6 years (range: 1–19). In addition, one patient (10%) demonstrated a 75% reduction in seizure frequency and two others (20%) had no significant effect on seizure frequency or severity. Cardiovascular follow-up (annually in the last 3 years of this analysis) showed no cases of PH. A slight thickening of one or two heart valves was demonstrated by two patients, but these changes were stable and not considered clinically significant by the cardiologist. Loss of appetite was reported in two patients, both on a combination therapy with TPM, another drug known to reduce the appetite [Klein et al. 2008]. Sleepiness and fatigue were, respectively, seen in one and two patients. Although two of them had a mild valve thickening on echocardiography, the valve abnormalities were not responsible for the fatigue. The fatigue was mainly reported at the start of the therapy and dose reduction or habituation to fenfluramine led to a disappearance of the fatigue.
Of interest was the response of six patients to a temporary withdrawal of fenfluramine after its removal from the market. Three patients who had been seizure-free during the previous year had a recurrence of seizures after withdrawal of fenfluramine and once again became seizure-free after restarting fenfluramine. Two other patients had no change in seizure frequency or severity during their withdrawal from fenfluramine, but their parents requested to restart fenfluramine when it became available due to fear of provoking status epilepticus.
The results of this study suggest that fenfluramine is effective against all seizure types, not only self-induced or photosensitive seizures.
Prospective data
In 2010, a prospective open label follow-up study of fenfluramine in patients with Dravet syndrome being treated under the Royal Decree was organized at Antwerp University Hospital (Edegem, Belgium). Patients from 6 months to 50 years of age with a diagnosis of Dravet syndrome were eligible to enroll. Patients with cardiovascular disease, including drug-treated hypertension and cardiac valvulopathy, were excluded. Efficacy and adverse events were monitored at scheduled and unscheduled office visits. FFA doses could be adjusted at the discretion of the treating physician up to a maximum of 20 mg/day. In 2012, 12 patients were included of which 10 from the retrospective study and two new patients. Of the seven of 10 patients from the retrospective study [Ceulemans et al. 2012] who had been seizure-free in the year prior to the initiation of this prospective study, four have continued to be free of seizures and three have exhibited rare seizure episodes (3–7 tonic–clonic convulsions) during the two additional years of observation. In three of them there was a provocative factor like fever, a stressful situation or intensive physical exercise explaining the seizures. Nevertheless seizures commonly fluctuate in patients with Dravet syndrome.
One of two patients who had been categorized as a nonresponder during the retrospective study demonstrated a 6-month seizure-free interval. An additional patient who had experienced a 75% reduction in seizure frequency after starting fenfluramine during the first year of treatment in the retrospective study continued to improve during the first 2 years of the prospective observation.
Two new patients with Dravet syndrome, aged 10.8 and 1.2 years at study entry, have enrolled in the study. At the start of fenfluramine they were, respectively, taking VPA+TPM+CLB and VPA+CLB. After starting fenfluramine (0.26 and 0.77 mg/kg/day, respectively) they both demonstrated a ⩾75% reduction in seizure frequency and disappearance of status epilepticus. Seizure-free periods were seen for 3 months in one child and 6 months in the other.
Cardiovascular follow-up does not show any serious or clinical relevant side effects. However long-term prospective data are currently being collected.
Discussion
The aim of this paper is to review the use of fenfluramine in the treatment of neurological disorders. Until now the literature is however sparse and almost limited to older case reports and small series. Only one retrospective trial has been published concerning the use of low-dose fenfluramine patients with Dravet syndrome [Ceulemans et al. 2012]. Since the use of fenfluramine is best demonstrated in self-induced seizures and Dravet syndrome we will focus hereon. Dravet syndrome is a rare epileptic encephalopathy estimated to affect one in 20,000 to 40,000 children [Hurst, 1990; Yakoub et al. 1992; Brunklaus et al. 2012]. In the last few decades, there has been substantial progress in understanding this devastating epilepsy syndrome which has led to a better approach and treatment of patients with Dravet syndrome. In Europe, either the combination of STP, VPA, and CLB or VPA in combination with TPM is used [Chiron and Dulac, 2011]. Although these combinations give acceptable results, many patients do not achieve adequate seizure control. Other treatment regimens include combinations with levetiracetam, clonazepam, and vagal nerve stimulators but often without good results. Clearly, better treatments are needed to address this patient population.
The retrospective data reported by Ceulemans and colleagues [Ceulemans et al. 2012] and summarized above suggest that fenfluramine may be an effective treatment for patients with Dravet syndrome. As noted above, fenfluramine appears to provide an antiepileptic action through a serotoninergic mechanism, making it unique among antiepileptic agents. However, the exact underlying mechanism of its anticonvulsant activity is still not known and interaction at other receptors cannot be ruled out at this time. It would be interesting to know whether specific 5-HT receptors are involved in the antiepileptic effect or not. With this knowledge more targeted and perhaps safer products could be developed in the future. The serious valvular heart disease risks associated with fenfluramine alone and in combination with phentermine [Abenhaim et al. 1996; Connolly et al. 1997] remain a major concern for any use of fenfluramine. Little evidence of clinically significant valvular changes were found in our retrospective analysis of patients with Dravet syndrome treated with fenfluramine. The low incidence of valvular changes may be the result of the relatively low doses used to treat Dravet syndrome (in all cases ⩽20 mg/day). In contrast, severe valvulopathy was more common in weight loss patients treated with fenfluramine at a dose ⩾60 mg/day than in patients taking ⩽40 mg/day [Li et al. 1999].
Conclusion
Fenfluramine is a promising AED in the treatment of Dravet syndrome. Given the severity of Dravet syndrome, the generally poor quality of life experienced by these patients, and our experience with years of use under the Belgium Royal Decree, we believe that fenfluramine exhibits a favorable benefit–risk profile in this patient population and offers a new and possibly effective treatment option. Further randomized controlled trials are planned for the near future as they are necessary to confirm our preliminary observations about the efficacy and the overall safety of this novel treatment when used in this patient population.
Footnotes
Acknowledgements
Professional medical writing was provided to the authors by Edward Weselcouch, PhD, of PharmaWrite, LLC (Princeton, NJ) and was paid for by Zogenix, Inc. This manuscript was prepared according to the International Society for Medical Publication Professionals’ ‘Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP2 Guidelines’ and the International Committee of Medical Journal Editors’ ‘Uniform Requirements for Manuscripts Submitted to Biomedical Journals’.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was sponsored by Zogenix, Inc. (Emeryville, CA). Editorial assistance was funded by Zogenix, Inc.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AS was a PhD student at the University of Antwerp and received an educational grant from Zogenix. BC and LL are members of the advisory board of Zogenix.