
Abstract
Acute and subacute inflammation, the mechanisms by which demyelination and axonal loss occur in multiple sclerosis (MS), result from the migration of activated immune cells into the central nervous system parenchyma. The triggering antigen is unknown, but the process involves deregulated immune response of T and B lymphocytes, macrophages, and mediators with expansion of autoreactive T cells creating a shift in the balance of pro- and anti-inflammatory cytokines favoring inflammation. Ongoing disease activity and exacerbations early in the course of relapsing–remitting MS may prevent full remission and propagate future progressive disability. A key strategy of immune therapy is timely initiation of treatment to achieve remission, followed by maintenance of remission. In this context, treatment with high-dose methylprednisolone (MP) is currently recommended to induce a faster recovery from a clinical exacerbation that results from an acute inflammatory attack. Adrenocorticotropic hormone (ACTH or corticotropin) gel is an alternative for patients who do not respond to or do not tolerate corticosteroids. ACTH is a universal agonist in the melanocortin (MC) system and, as such, among other functions, stimulates the adrenal cortex to produce cortisol. MCs are a family of peptides that includes ACTH and other MC peptides. This system has five classes of receptors, all of which show a strong affinity for ACTH, suggesting a more complex and dynamic mechanism than only inducing endogenous corticosteroid production. ACTH and MCs regulate processes relevant to MS, including anti-inflammatory and immunomodulatory functions involving lymphocytes, macrophages, the sympathetic nervous system involved in inflammatory processes, and reduction of pro-inflammatory cytokines. The clinical implications of the mechanistic differences between corticosteroid and ACTH gel treatment remain to be elucidated. Recent data show that patients experiencing an acute exacerbation, who previously had suboptimal response to or were unable to tolerate MP treatment, showed positive clinical outcomes with fewer adverse events with ACTH gel.
Keywords
Background
Relapsing forms of multiple sclerosis (MS) are prevalent clinical types of MS and include relapsing–remitting MS (RRMS), relapsing forms of secondary-progressive MS (SPMS) and progressive–relapsing MS (PRMS); MS relapse is a clinical and immunological hallmark of these conditions. Relapsing MS is characterized by recurrent, persistent, and chronic inflammation of the central nervous system (CNS) which manifests clinically with recurrent episodes of new or worsened symptoms. Acute episodes of neurological dysfunction (exacerbations) are followed by periods in which there may be partial or complete remission, with apparent clinical stability between relapses [Confavreux et al. 2000]. The exacerbations may occur over days or even weeks with prolonged periods of functional recovery that may last months [Goodin et al. 2002]. Between exacerbations, patients tend to be neurologically and symptomatically stable, although neurological disability may result from relapses that have incomplete remission [Goodin et al. 2002]. Frequent relapses in the first 2 years after diagnosis have been associated with later disability, reflected by an increased probability of entering the secondary progressive phase [Scalfari et al. 2010]. In addition, incomplete recovery from relapse appears to be associated with sustained disability progression. Analysis of data from the Natalizumab Safety and Efficacy in Relapsing Remitting Multiple Sclerosis (AFFIRM) trial and the Multiple Sclerosis Collaborative Research Group (MSCRG) indicated that more patients had sustained progression due to incomplete recovery from relapses than sustained progression without relapses [Scott et al. 2013].
An exacerbation is defined as symptoms occurring over a minimum of 24 hours that follow a previous attack by at least 30 days [Frohman et al. 2008; Ontaneda and Rae-Grant, 2009], in the absence of fever or infection. Inflammatory events with demyelination and neuronal and axonal loss are believed to accumulate over time and manifest as clinical deterioration, leading to disability and a reduced quality of life [Lublin et al. 2003; Berkovich, 2013]. However, each exacerbation may manifest differently, with a wide range of symptoms and severity as a function of the location and strength of the inflammatory insult (Table 1) [Catania et al. 2004; Frohman et al. 2008; Berkovich, 2013]. A temporary reactivation of existing symptoms (pseudoexacerbation) or temporary neurological dysfunction (paroxysmal symptom) is often a result of a temperature-dependent conduction block in demyelinated axons, triggered by an increase in body temperature [Berkovich, 2013]. Unlike a bona fide exacerbation, a pseudoexacerbation is a temporary aggravation of existing symptoms, often triggered by a precipitating agent, and typically shows a temporal relationship between the trigger and the symptoms rather than a true relapse of the disease itself, which is distinct in time [Sibley et al. 1985; Repovic and Lublin, 2011]. Paroxysmal symptoms differ from disease exacerbations or pseudoexacerbations in that they are fleeting neurological disturbances, such as paresthesias or spasms, and often persist for only seconds to minutes [Schapiro, 2001]. These symptoms will often resolve spontaneously with cooling; however, the offending agent must be identified for treatment before the emergent symptoms can be considered a bona fide exacerbation [Sibley et al. 1985].
Sites of MCR expression and neurobiologic activity potentially relevant to MS.

ACTH, adrenocorticotropic hormone; MCR, melanocortin receptor; MS, multiple sclerosis; MSH, melanocyte stimulating hormones.
Adapted from Catania et al. [2004]. Reprinted with permission.
For relapses that are severe or disabling enough to require treatment, high-dose corticosteroids are generally used as first-line treatment. Adrenocorticotropic hormone (ACTH; H.P. Acthar® Gel, repository corticotropin injection; Questcor Pharmaceuticals, Inc.), a long-acting formulation of the full sequence ACTH(1-39) (80 units/ml) that may include other pro-opiomelanocortin (POMC) peptides, is approved by the US Food and Drug Administration for treatment of MS relapses [Questcor Pharmaceuticals, 2012]. This review focuses on the use of ACTH gel for the treatment of acute exacerbations of RRMS, with particular emphasis on the mechanistic rationale.
Immunobiology of acute multiple sclerosis exacerbations
Whereas chronic inflammation in the CNS plays a role in MS-associated progression and disability, acute inflammatory exacerbations are the mechanism by which demyelination and axonal loss are believed to occur [Noseworthy et al. 2000; Lublin et al. 2003; Frohman et al. 2005; Berkovich, 2013]. The activation of the immune process is initiated systematically, resulting in migration of activated immune cells into the CNS where they are reactivated and result in parenchymal inflammation [Markovic-Plese et al. 2004].
The inflammation in MS may be focal, multifocal, or diffuse and is characterized by infiltration of activated lymphocytes, macrophages, and microglia, with involvement of cortex, white matter, and deep gray matter with myelin destruction, axonal, neuronal, and synaptic loss, astroglial reaction, remyelination, and synaptic rearrangement [Frohman et al. 2006; Lassmann et al. 2007].
Although the antigen/trigger remains unknown in MS, a deregulated immune response, including inflammatory cells (e.g. T cells, B cells, macrophages) and mediators [e.g. cytokines, chemokines, matrix metalloproteinases (MMPs), complement], contributes to the expansion of autoreactive T cells [Boppana et al. 2011]. Defunct regulatory T cells (Tregs) enhance the expansion of autoreactive pro-inflammatory cells [Boppana et al. 2011]. Pro-inflammatory cells express very late antigen-4 (VLA-4) that binds to vascular cell adhesion molecule-1 (VCAM-1) and secrete MMPs, allowing blood–brain barrier lymphocyte and monocyte extravasation [Wilson et al. 2010]. Various T helper cell responses (i.e. helper T type 1 [Th1, Th2, Th3, Th17], type 1 T regulatory [Tr1] or Treg) may be induced, based on the cytokine milieu [Fletcher et al. 2010].
Th1 cells historically have been thought to be the main effector T cells responsible for immune-mediated inflammation [Fletcher et al. 2010]. Th1 cells produce tumor necrosis factor gamma (TNF-γ)-mediated inflammation of the CNS in MS and experimental autoimmune encephalomyelitis (EAE) pathogenesis, the latter being an animal model of inflammatory, demyelinating disease of the CNS; whereas Th2 cells have a beneficial effect on the disease [Rostami and Ciric, 2013]. The Th1/Th2 paradigm remained the prevailing view of MS/EAE until 2005 when a new lineage (Th17) was discovered and shown to play an essential role in many inflammatory diseases, including EAE as well as MS, and appears to be important in the initial phases of disease [Steinman, 2007; Rostami and Ciric, 2013].
Whereas the role of Th1 cells is considered contributory in the pathogenesis, perhaps more so later in the disease, the current view on the role of Th cells in MS/EAE pathogenesis is characterized by the Th1/Th17 paradigm. Although the precise role of a Th1/Th17-mediated response in MS remains unclear, the discovery that αB-crystallin is produced in copious amounts in MS lesions suggests that it is a guardian molecule because it attenuates disease and downregulates the production of Th1 and Th17 pathways and binds these pro-inflammatory mediators in plasma [Fletcher et al. 2010; Steinman et al. 2013]. In this context a systemic humoral response to αB-crystallin correlates with disease activity and severity [Agius et al. 1999].
Tregs normally control the intensity of an immune response; however, research to elucidate their regulatory function in MS exacerbations needs more intensive study [Hollifield et al. 2003; Correale and Villa, 2010]. Accumulating evidence indicates an immunosuppressive role for CD4+ CD25+ Tregs in immune-mediated diseases. Venken and colleagues suggested that the suppressive capacity of these Tregs appears to be more affected in the early phase of the disease process [Venken et al. 2006]. Consistent with this, there are differences in function and expression of FOXP3 (a master regulator in the development and function of regulatory T cells) of CD4+ CD25+ T cells, which are adequate in patients with SPMS and reduced in those with RRMS [Venken et al. 2006]. More recently, Correale and Villa investigated the role of CD8+ CD25+ FOXP3 in RRMS patients, as the role of CD8+ has received less attention than CD4+ [Correale and Villa, 2010]. CD8+ CD25+ FOXP3 is suppressed to a greater degree in the peripheral blood during MS exacerbations than during remission or in healthy controls [Correale and Villa, 2010]. Likewise, in the cerebrospinal fluid (CSF) of MS patients during exacerbations, lower levels of CD8+ CD25+ FOXP3 T cells are detected, providing evidence for a previously unknown mechanism involved in immune regulation in MS [Correale and Villa, 2010].
Boppana and colleagues reviewed the immunologic aspects of MS [Boppana et al. 2011]. Although autoreactive T-cell-mediated immune response is considered critical for MS pathogenesis, there is increasing evidence that B lymphocytes also play a key role [Boppana et al. 2011]. The recent findings of B-cell lymphoid follicles found in the meninges of MS patients with progressive disease supports an important role for B cells in the pathogenesis of MS. Meningeal follicles are thought to play a role in the formation of cortical lesions in MS as well as gray matter atrophy [Boppana et al. 2011]. The pro-inflammatory cytokine shift in patients with MS demonstrates a decrease in anti-inflammatory cytokines including interleukin (IL)-10; IL-10-producing B cells are reported to be deficient in MS (Figure 1) [Boppana et al. 2011].

Cytokine imbalance in MS. The healthy immune system maintains a balance between pro- and anti-inflammatory cytokines, whereas individuals with MS have an imbalance of cytokines related to increased Th1 immune response and decrease in Th2/Treg function characterized by increased expression of pro-inflammatory cytokines IFN-γ, IL-12, TNF, and IL-17.
The cells of the innate immune system play an important role in the initiation and progression of MS by influencing the T- and B-cell effector function, which alters the expression and balance between pro- and anti-inflammatory cytokines and thus neural inflammation [Breij et al. 2008; Gandhi et al. 2010; Boppana et al. 2011]. These cells include dendritic cells (DCs), microglial cells/macrophages, natural killer (NK) cells, mast cells, natural killer T cells (NK-T), gamma-delta cells (GD-T) and complement activation [Breij et al. 2008; Gandhi et al. 2010].
Dendritic cells
Dendritic cells are antigen presenting cells (APCs) that interact with T cells to initiate and shape the adaptive immune response [Gandhi et al. 2010]. The interaction between DCs and T cells is important for differentiating T cells into either effector T cells such as Th1, Th2, and Th17 or Tregs such as Tr1 [Gandhi et al. 2010]. In the experimental model of EAE pathogenesis, DCs accumulate in the CNS during inflammation and promote EAE pathology through the secretion of pro-inflammatory cytokines and activation markers. Involvement of DCs in the EAE model may be relevant to MS in that studies show DCs have an activated phenotype with expression of activation markers and aberrant secretion of pro-inflammatory cytokines [Gandhi et al. 2010]. In EAE, DCs also secrete a glycoprotein (osteopontin) that is involved in chemotaxis, activation, and differentiation of immune cells and shows increased expression in brain lesions during EAE [Gandhi et al. 2010].
Microglial cells
Microglia are considered resident macrophages in the CNS and, in addition to phagocytosis, they contribute to MS and EAE pathology through antigen presentation and secretion of pro-inflammatory cytokines [Gandhi et al. 2010]. The process of antigen presentation by microglia involves a family of molecules found only in APCs and lymphocytes called major histocompatibility complex (MHC) class II and the costimulatory molecules CD83 and CD40, which are required for the interaction of effector T cells and B cells [Gandhi et al. 2010]. Microglia are involved in demyelination and phagocytosis of degraded myelin. As a consequence of myelin degradation, reactive oxygen species and myeloperoxidases are expressed and contribute to neuronal damage. Microglia can also promote cell death by augmenting the secretion of an apoptotic inflammatory cytokine (TWEAK) [Gandhi et al. 2010].
Natural killer cells
A primary role of NK cells relates to their cytotoxic activity against viral infection [Gandhi et al. 2010]. The role that NKs play in the pathophysiology of immune-mediated disease is unclear. NK cells are thought to interact with DCs during the early stages of immune response by stimulating DC maturation and increasing the amount of DC-produced cytokines. It is also thought that NKs, or certain subtypes of NKs, may play a regulatory role in MS by modulating the activation and survival of autoreactive T cells and MCs through cytokine production and cytotoxicity [Gandhi et al. 2010].
Mast cells
Mast cells are present in the normal brain in the parenchyma and at the blood–brain barrier and can interact with myelin and accumulate in the border zones of MS plaques [Gandhi et al. 2010]. The observation of elevated mast cells tryptase in CSF supports the hypothesis that mast cells may be a functional element at the onset and maintenance of the MS disease process, particularly in females where the difference in mast cells between males and females is significant and of interest because females are known to be more inclined to develop MS than males [Gandhi et al. 2010; Krüger and Mørk, 2012].
Natural killer T cells
Much has yet to be learned about NK-T cells, which are a subset of NK and T cells [Gandhi et al. 2010]. NK-T cells have been linked to inflammation and immune-mediated disease through the modulation of anti-inflammatory Th2 cytokines such as IL-4, IL-10, and tumor growth factor (TGF)-β, and may be involved in the remission phase of MS [Gandhi et al. 2010; Boppana et al. 2011]. NK-T cells can also produce IL-10, which is shown to be essential for induction of Tregs [Gandhi et al. 2010].
Gamma-Delta T cells
GD-T cells are a subset of lymphocytes and their precise role in MS is unclear [Gandhi et al. 2010]. For example, in EAE, these cells have been shown to either aggravate EAE or show no effect. However, GD-T cells are present in MS lesions and increase in MS patients with active disease and are present in the clinical context of high magnetic resonance imaging (MRI) activity [Gandhi et al. 2010].
Complement activation
Complement activation has long been suspected to play a pathological role based on the results of pathological and functional studies, which have demonstrated complement activation products in MS brain and biological fluids. However, the extent and nature of complement activation and its contribution to disease phenotype and long-term outcome remain unclear. A post-mortem study showed that complement and antibodies were consistently associated with macrophages in areas of active demyelination in established MS [Breij et al. 2008]. There was evidence that the initial apparent heterogeneity of demyelinating lesions in the earliest phase of MS lesion formation may disappear over time, suggesting that different pathways may converge in one general mechanism of demyelination [Breij et al. 2008]. Immunoglobulin G (IgG) antibodies were always associated with active demyelination. Thus, the consistent presence of complement, antibodies and Fcγ receptors in phagocytic macrophages suggests that complement- and antibody-mediated myelin phagocytosis may be the dominant mechanism of demyelination in established MS [Breij et al. 2008].
Remyelination is a spontaneous process that appears to occur sometimes remarkably in adults with MS and seems to occur when oligodendritic progenitor cells (OPCs) respond to axonal injury and differentiate into oligodendrocytes [Franklin et al. 2012]. Incomplete remyelination following exacerbations accumulates over time and contributes to clinical deterioration [Lublin et al. 2003; Berkovich, 2013]. Mechanisms proposed for axonal damage are many and varied [Franklin et al. 2012]. Avoiding the formation of new inflammatory lesions may be expected to be of benefit by preventing axonal transection that accompanies the formation of these lesions in MS [Franklin et al. 2012]. Oxidative damage is important in MS lesions with the generation of reactive oxygen species that accompany myelin degradation and could mediate mitochondrial damage [Franklin et al. 2012]. In addition, adenosine triphosphate (ATP) powers the ATPase sodium pump and the accumulation of sodium and membrane depolarization may contribute to axonal damage [Franklin et al. 2012]. Further, astroglial scarring impairs remyelination and repair, increasing disability [Nair et al. 2008].
Multiple sclerosis exacerbations and progressive disability
Ongoing disease activity and exacerbations, reflecting CNS inflammation and demyelination early in the course of MS, may propagate future progressive disability [Confavreux et al. 2003; Berkovich, 2013]. Gender, age, initial symptoms, course of disease, degree of recovery from first exacerbation, the number of relapses occurring over the first 5 years of the disease, the time from original onset to a second neurological symptom, and a score of 4 on the Kurtzke Disability Status Scale are strong predictors of future disability [Kurtzke, 1983; Confavreux et al. 2003]. Whereas residual disability accumulates with each exacerbation [Lublin et al. 2003; Scalfari et al. 2011], reducing the number of CNS lesions by means of disease-modifying therapy early in the course of MS may delay or reduce the risk of disability [Weinshenker et al. 1989; Tremlett et al. 2009; Rudick and Kappos, 2010]. Indeed, remission at onset of disease followed by maintenance of disease remission is a strategy to prevent long-term disability in chronic immune-mediated disorders [Richman and Agius, 2003]. Long-term studies of interferon-beta (IFN-β) therapy indicate that delayed or discontinued treatment provides less benefit than continuous therapy [Carroll, 2009]. Observations from the Betaferon®/Betaseron® in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) and Prevention of Relapses and Disability by Interferon-β-1a Subcutaneously in Multiple Sclerosis (PRISMS) trials show a prolongation in time to confirmed disability progression [PRISMS Study Group and University of British Columbia MS/MRI Analysis Group, 2001; Kappos et al. 2009]. Worsening of the Expanded Disability Status Scale (EDSS) 1.0 or more points has been a common definition of disability progression. EDSS worsening of more than 1 point from baseline during the Avonex® pivotal trial, correlated with increased risk of disability at 8 years [Rudick et al. 2010]. Thus, all patients with a diagnosis of definite RRMS should begin disease-modifying therapy at the earliest opportunity, as these agents are not only more effective in treating RRMS than SPMS, but also appear more effective in early RRMS. In this way, early intervention may improve long-term outcomes [Goodin, 2008].
MRI T2 and gadolinium (Gd)-enhanced T1 lesions correlate with development of disability [Rio et al. 2008; Sormani et al. 2009, 2010]. Currently available medications that target the inflammatory component of MS show reduction in the accumulation of T2 burden of disease and the numbers of Gd-enhanced lesions. Thus, disease activity as measured by MRI T2 volume is an important variable in determining response to preventive immune therapy and future dis-ability [Prosperini et al. 2012]. Furthermore, Gd-enhanced T1-weighted lesions represent a reliable and sensitive biomarker of active new lesions and inflammation in RRMS. They correlate with the number of relapses [Rovaris et al. 2003], and, in patients receiving preventive immune therapy, they are also a variable determining future disease activity and future disability [Rio et al. 2008, 2009].
In contrast, in individual patients with MS, relapse symptoms often do not correlate well with MRI changes. They may occur before MRI changes are evident or symptoms may be caused by pathologic damage with a clinical expression that may not be readily detectable with conventional MRI [Sormani et al. 2009]. For example, two of the most common symptoms of MS are chronic fatigue and pain, and these symptoms bear little relationship with MRI changes [Chaudhuri and Behan, 2005]. However, a strong correlation has been shown between the total number of sensory symptoms reported and the presence of disability [Rae-Grant et al. 1999]. Thus, in the practice setting, the diagnostic process is typically triggered by the presentation of new or worsening relapse symptoms, and the decision to initiate acute treatment for relapse is often made without confirmation of new disease activity by MRI.
Conversely, Gd-enhanced lesions may be detected in the course of a routine MRI, while the patient does not exhibit obvious symptoms of a relapse. Initiation of relapse treatment for Gd-enhanced lesions in this context has not been investigated in clinical trials and remains a matter of debate [IFNB Multiple Sclerosis Study Group and University of British Columbia MS/MRI Analysis Group, 1995; PRISMS Study Group and University of British Columbia MS/MRI Analysis Group, 2001] At the same time, in clinical trials, Gd-enhanced lesions are frequently used as a radiologic equivalent of MS relapse both in the context of inclusion criteria and as a measure of treatment effectiveness [Gold et al. 2012; Comi et al. 2013; Lublin et al. 2013]. As such, detection of a Gd-enhanced lesion indicates the presence of active inflammatory activity and may warrant acute treatment even if the relapse presentation is not obvious clinically. Indeed, the aforementioned evidence that inflammatory demyelinating events contribute to disability suggests that, in this context, there may be a longer-term benefit to initiating relapse treatment and to inducing and maintaining a remission.
Cognitive disturbances are a prominent feature of MS and are estimated to occur in one half of patients [Strober et al. 2009]. Cognitive impairment often accompanies or follows acute relapses [Foong et al. 1998; Morrow et al. 2011]. While some evidence suggests that reduction of Gd-enhanced lesions is associated with improvements in cognitive symptoms [Foong et al. 1998], there is currently little information available regarding potential benefits of relapse treatment for cognitive relapses (i.e. acute cognitive impairment that is not accompanied by other relapse symptoms). As relapsing MS progresses with acute attacks, cognitive impairment may become more apparent, as a longitudinal analysis shows that cortical lesion increase over time is associated with cognitive impairment, particularly in SPMS patients [Rao et al. 1991; Roosendaal et al. 2009]. A cross-sectional analysis showed that cognitively impaired patients had significantly higher cortical lesions and total lesion volume compared with cognitively unimpaired patients [Calabrese et al. 2009]. Another study showed that baseline cognitive deficits related to processing speed and verbal memory were correlated significantly with EDSS change over 5–7 years later [Deloire et al. 2010]. Acute cognitive changes need further attention to better characterize the relationship with relapse and MRI evidence of active inflammatory disease, as well as appropriate management of cognitive relapses.
Treatment options
Acute therapy
The initial strategy is early, proactive immune treatment of MS, which is supported by evidence of a prominent inflammatory immune response that is more marked early in the course of the disease. Relative to placebo or no treatment, high-dose corticosteroids (CS) or ACTH produce more rapid and robust recoveries during exacerbations [Rose et al. 1970; Thompson et al. 1989; Sellebjerg et al. 1998]. Treatment with high-dose methylprednisolone (MP) has been shown to induce a faster recovery from an acute attack [Sellebjerg et al. 2005]. Guidelines from the European Federation of Neurological Societies (EFNS) for the treatment of MS relapses currently recommend high-dose MP for the acute treatment of relapses (Level A) [Sellebjerg et al. 2005]. However, the optimal regimen in terms of efficacy and adverse effects remains to be established in randomized, controlled trials [Sellebjerg et al. 2005]. Sellebjerg and colleagues demonstrated a significant improvement on EDSS score in MP-treated patients compared with placebo [Sellebjerg et al. 1998]. Self-reported data from the North American Research Committee on Multiple Sclerosis (NARCOMS) showed that a substantial percentage of patients reported that treatment with either intravenous methylprednisolone (IVMP) or oral corticosteroids resulted in no change or incomplete recovery (30% IVMP and 38% oral corticosteroids) [Nickerson and Marrie, 2013]. Corticosteroid failure may be considered if a patient fails to return to their previous level of functioning after acute treatment of exacerbation with steroid. However, residual deficit after relapse may persist and contribute to stepwise progression of disability [Berkovich, 2013]. On an objective basis, pooled placebo data from clinical trials indicate that up to one-half of untreated patients may have post relapse residual disability of at least 0.5 EDSS and about one-third of patients may exhibit residual disability of at least one point EDSS [Lublin et al. 2003; Hirst et al. 2008]. Acute intervention with MP or ACTH gel can be expected to demonstrate less residual disability than shown in the pooled placebo data and failure to achieve EDSS < 0.5 provides an objective basis for treatment failure.
ACTH gel is an alternative to high-dose corticosteroids in the treatment of acute exacerbations in patients who do not respond to or do not tolerate CS [Ross et al. 2013]. In the experience of one of the authors (RB), at least 55–60% of patients who are steroid failures may respond to ACTH gel in different degrees. This alternative consideration is based on a systematic review of ACTH or CS in six randomized, controlled trials to primarily determine whether ACTH or CS reduced short-term disability after an exacerbation [Filippini et al. 2000]. Overall, ACTH or MP showed a protective effect against worsening or stabilizing MS. Short-term administration of 5–15 days did not show any significant difference between treatments. ACTH gel or MP therapy was associated with a significant reduction in the risk of being worse or not improved within 5 weeks (odds ratio [OR] = 0.37, 95% confidence interval [CI] 0.24–0.57). In absolute terms, this means that 247 more patients improved for every 1000 patients treated with corticosteroids or ACTH gel (95% CI 144–349, improved) [Filippini et al. 2000].
Although not relevant to the question of acute relapse treatment, the use of pulse or chronic use of MP is often raised when considering the use of CS in MS because of evidence that prolonged treatment may slow the rate of brain atrophy and development of T1 black holes [Zivadinov et al. 2001]. The effect of chronic administration of MP or ACTH on progression is unclear [Ellison and Myers, 1980]. A Cochrane review of the long-term efficacy and safety of CS therapy showed that there is no evidence that long-term CS treatment delays progression of long-term disability [Ciccone et al. 2008]. One study showed that high-dose intravenous MP (IVMP) was associated with a significant reduction in disability risk at 5 years, but it was considered to be at high risk of bias [Ciccone et al. 2008]. Much less is known about the potential effects of chronic administration of ACTH. Thus, this question remains open until such time that a randomized, adequately powered, high-quality study is done to provide a better understanding of the risk versus benefit of chronic dosing of IVMP and ACTH; however, this has little relation to the question of acute relapse treatment.
Plasmapheresis is mainly used in severe relapses that are refractory to high-dose CS and ACTH gel, and published data on its efficacy are limited [Magana et al. 2011; Ross et al. 2013]. Off-label use of intravenous immunoglobulin (IVIG) for MS relapses is not supported by robust evidence [Gray et al. 2003].
ACTH gel mechanism of action
ACTH is one of several physiologically active peptides synthesized in vivo by proteolytic cleavage of the precursor peptide pre-POMC. ACTH is a universal agonist for the melanocortin receptor (MCR) system, which is a family of peptides that includes ACTH (corticotropin) and other melanotropins designated as α-, β-, γ-melanocyte stimulating hormones (MSHs); as such ACTH, among other functions, also stimulates the adrenal cortex to produce cortisol in response to stress. This function of ACTH is better studied and known and, until relatively recent times, was viewed as the only mechanism and function of ACTH [Gong, 2012].
Outside the United States, a synthetic form of ACTH (Synacthen®) is available that contains a peptide comprising the first 24 of 39 amino acids that constitute naturally occurring ACTH [Gong, 2012]. In the United States, a proprietary mixture of porcine pituitary extracts (ACTH gel) contains the 39-amino acid peptide ACTH and other potentially bioactive POMC peptides. A growing body of evidence suggests that naturally occurring ACTH differs from synthetic analogues, not only in terms of its structure, but also its pharmacokinetics and bioactivity [Gong, 2012].
The effectiveness of ACTH gel in treatment of acute exacerbations of MS in patients who have failed CS may be related to its mechanism of action. The MCR system has five MC receptors (MC1R-MC5R), all of which show a strong affinity for ACTH, suggesting a more complex and dynamic mechanism than solely inducing endogenous corticotropin production [Arnason et al. 2013]. Indeed, ACTH only enhances endogenous CS production through binding with MC2R. At the same time, ACTH binding to other MCRs can regulate other processes that are potentially relevant to MS such as anti-inflammatory and immunomodulatory functions involving T and B lymphocytes, macrophages, sympathetic nervous system involved in inflammatory processes, reduction of pro-inflammatory cytokines, inflammatory nitric oxide, adhesion molecules, production of anti-inflammatory IL-10 for which IL-10 producing B cells are reported to be deficient in MS, as mentioned [Arnason et al. 2013].
MC1R expression occurs in macrophage/monocyte cells and glial cells [Catania et al. 2004; Ross et al. 2013]. Transactivation of MC1R in inflammatory cells causes marked reduction of the activation and translocation to the nucleus of transcription factor (NF-κB) [Catania et al. 2004]. This could be an important factor underlying the anti-inflammatory effects of MC1R, as it regulates the expression of several genes in the immune/inflammatory process, including those that code for pro-inflammatory cytokines, MMP, and adhesion molecules [Ross et al. 2013]. ACTH reduces pro-inflammatory cytokines through the modulation of NF-κB signaling, as marked anti-inflammatory effects are exerted through inhibition of NF-κB [Catania et al. 2004].
MC2R is prevalent in the adrenal cortex and mediates the production and release of steroids by the adrenal cortex, circadian rhythm, and stress-related fluctuations [Catania et al. 2004]. MC2R is unique since only ACTH will activate this biologic receptor [Getting, 2006].
MC3R expression in the brain obtained by in situ hybridization is abundant in the hypothalamus and limbic system, but signals for this receptor are also present in the septum, thalamus, hippocampus, and midbrain [Catania et al. 2004]. It appears to participate in autonomic functions, feeding, and activation of MC3R has clear anti-inflammatory influences, with MC3R receptors detected in peritoneal macrophage and knee joint macrophage [Catania et al. 2004; Getting, 2006]. Activation in the heart is shown to have protective effects associated with ischemic-reperfusion injury, and it is proposed that MC3R is an active modulator of the mechanism operating in inflammation [Getting, 2006].
MC4R is present in the CNS and is expressed within many regions, including MC4R mRNA-containing cells located in the anterior, central, and posterior hypothalamus, the dorsal horn of the spinal cord, and motor areas of the cortex; and serves in the regulation of energy homeostasis [Mountjoy et al. 1994]. MC4R may be of most interest regarding its role in food intake and energy expenditure [Catania et al. 2004; Getting, 2006].
MC5R is uniquely, extensively expressed in a variety of peripheral tissues, including bone marrow (a nest for migratory memory T cells), thymus and liver (lymphocyte enrichment in NK and NK-T cells that may have crucial roles in the recruitment of circulating T cells), and lymph nodes (T cell priming by DCs) [Crispe, 2003; Mempel et al. 2004]. Another interesting observation about MC5R is in its expression in B and T lymphocytes, which suggests the possibility of its participation in immune regulation [Catania et al. 2004; Getting, 2006]. MC5R induces Treg development, decreasing the recognition of self-antigen [Taylor and Namba, 2001].
MCR activation can induce anti-inflammatory signals via sympathetic/adrenergic pathways. This entails modulation of neurotransmitters that may occur through the release of norepinephrine and reduced production of pro-inflammatory cytokines by monocytes/macrophages [Schorr and Arnason, 1999]. α-MSH can induce DC25+ CD4+ Tregs through the MC5R expressed on primed T cells [Catania et al. 2004].
The clinical implications of the mechanistic differences between CS and ACTH gel treatment remain to be elucidated. However, what is clear is that MCR activation causes a collective reduction of molecules involved in inflammatory processes [Catania et al. 2004]. The extent to which this is meaningful needs further study in the clinical setting, as it may help to explain recent observations.
One recent study showed that patients experiencing an acute exacerbation of their MS, who previously failed MP treatment, demonstrated positive clinical outcomes with fewer adverse events with ACTH gel treatment [Berkovich et al. 2012]. This retrospective study evaluated the efficacy and safety of ACTH gel in reducing short- and long-term morbidity associated with MS relapses in 18 patients with RRMS who had previously failed steroid treatment [Berkovich et al. 2012]. All patients had failed IVMP 1 G × 3–5 days (17 patients) or oral MP 1 G × 7–10 days (two patients); two patients had previously been treated with ACTH. Patients received treatment for relapse with ACTH gel 80 USP units IM or SC once daily for 5 or 7 days. Their clinical status was then rated categorically as worsening, no change, minimal change, good, very good, or excellent. Previous treatments and outcomes are summarized in Table 2. After treatment with ACTH, the patients who previously failed MP treatment experienced positive clinical outcomes (Figure 2). None of the adverse events previously reported with MP were reported after ACTH gel; there was one report of mild lower extremities edema while on ACTH gel, which resolved after the treatment was completed [Berkovich et al. 2012]. Thus, further study is warranted for more than exploring potential differences in clinical outcomes; deeper exploration of the underlying mechanisms may provide additional insights into the MS disease process.
Summary of demographic, clinical characteristics, prior treatment, and reasons for failed MP treatment.
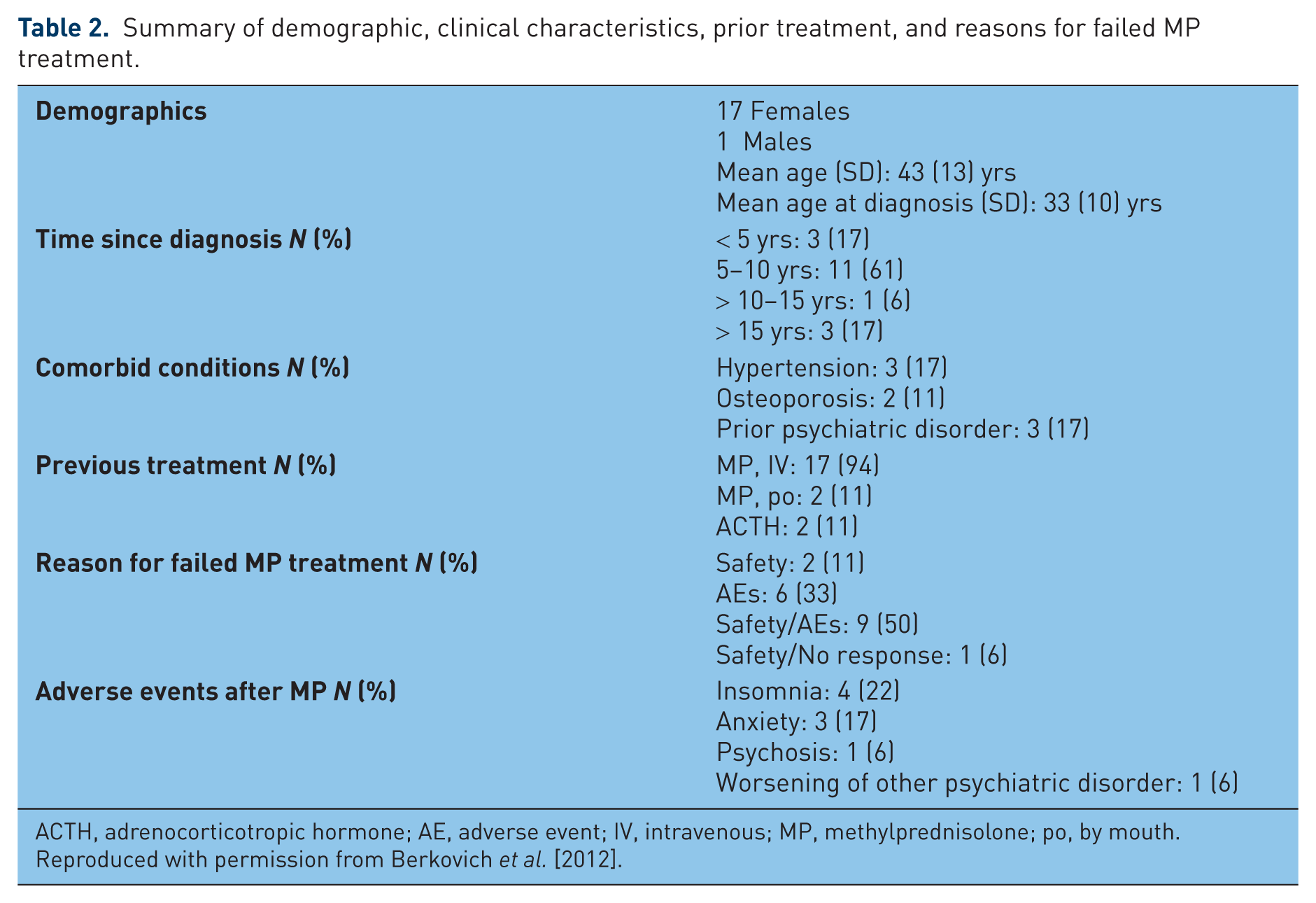
ACTH, adrenocorticotropic hormone; AE, adverse event; IV, intravenous; MP, methylprednisolone; po, by mouth.
Reproduced with permission from Berkovich et al. [2012].
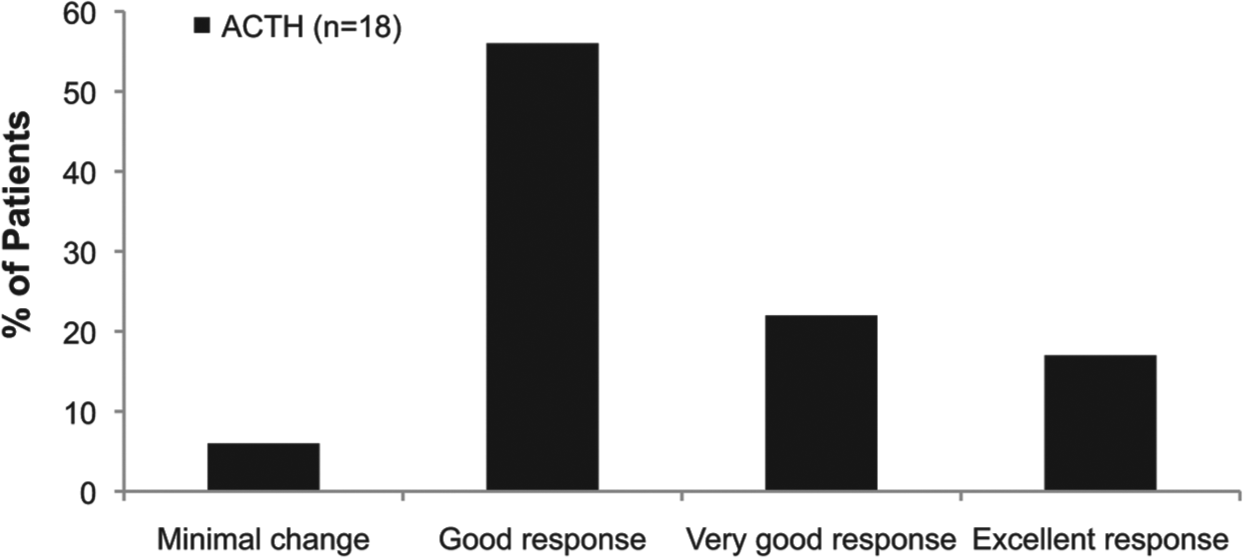
Physician-reported response of patients to ACTH after failing treatment with MP. Categorization of response is based on clinical evaluation of each patient after treatment with ACTH.
Summary
The emergent paradigm in the management of MS is toward early intervention with immunotherapeutic agents that target inflammation. Limited data show that patients who previously failed MP or experience breakthrough MS may benefit from ACTH gel. The complex and dynamic mechanism of action of ACTH through the MCR system has expanded the understanding of the role it plays in treatment of MS exacerbations and opens new horizons for therapeutic management of the disease process in MS. In this way, reducing disease activity and exacerbations early in the course of MS may delay and alleviate the future potential of progressive disability in individual patients.
Footnotes
Funding
Medical writing and editorial support was provided by Frank Beebe, PhD, of MedVal Scientific Information Services, LLC, and funded by Questcor Pharmaceuticals, Inc.
Conflict of interest statement
RB has served on advisory boards for Acorda, Avanir, Bayer, Biogen Idec, Genzyme, Teva, and Questcor; she has served as speaker for Acorda, Avanir, Bayer, Biogen, Genzyme, and Teva; and she has received financial support for investigator-initiated studies from National MS Society, Questcor, and Teva. MA has received personal compensation for activities with Teva Neuroscience, Biogen Idec, Berlex Labs, and Serono, Inc. and has received research support from Novartis, Teva Neuroscience, Biogen Idec, Genzyme Corporation, Roche Diagnostics Corporation, Immune Tolerance Network, Actelion, and Acorda Therapeutics.