
Abstract
Pharmacological targeting of ion channels has long been recognized as an attractive strategy for the treatment of various diseases. Multiple sclerosis (MS) is an autoimmune disorder of the central nervous system with a prominent neurodegenerative component. A multitude of different cell types are involved in the complex pathophysiology of this disorder, including cells of the immune system (e.g. T and B lymphocytes and microglia), the neurovascular unit (e.g. endothelial cells and astrocytes) and the central nervous system (e.g. astrocytes and neurons). The pleiotropic expression and function of ion channels gives rise to the attractive opportunity of targeting different players and pathophysiological aspects of MS by the modulation of ion channel function in a cell-type and context-specific manner. We discuss the emerging knowledge about ion channels in the context of autoimmune neuroinflammation. While some pharmacological targets are at the edge of clinical translation, others have only recently been discovered and are still under investigation. Special focus is given to those candidates that could be attractive novel targets for future therapeutic approaches in neuroimmune autoinflammation.
Introduction
Owing to their ubiquitous expression throughout all tissues and their essential importance for cellular functions, pharmacological targeting of ion channels opens up the possibility of cell-type and context-specific modulation of both immunological and neurodegenerative mechanisms in the context of autoimmune neuroinflammation (Figure 1). However, the broad expression pattern of most ion channels and their partial functional redundancy together with a lack of highly specific pharmacological inhibitors makes it difficult to target specific channel subtypes, leading to some limitations of ion channel modulation in vivo. Some of these ion channels (e.g. potassium channels on T cells and sodium channels on neurons) can be regarded as established pharmacological targets, while clarification of the targeting of other cell types and channel families is mostly still in the hands of basic researchers. Because of the vast amount of data on the topic, it is not possible to describe all the cell types and ion channels in detail here. We refer the reader to other in-depth reviews for additional information (e.g. monocytes/macrophages/microglia [Skaper, 2011; Waxman, 2006], oligodendrocytes [Matute, 2008] or endothelial cells [Nilius and Droogmans, 2001]).

Pathophysiology of multiple sclerosis. In lymphoid organs autoreactive T cells interact with antigen-presenting cells and B cells and, after activation, are able to cross the blood–brain barrier. In the central nervous system, reactivation of autoreactive T cells results in production of effector cytokines such as interferon γ, tumour necrosis factor α and interleukin 17, attraction of macrophages and microglia, antibody production by plasma cells and attack by CD8+ T cells. Together these mechanisms lead to demyelination and axonal injury. Cells, grey: oligodendrocytes; yellow: neurons; green: T cells; blue: macrophages; orange: astrocytes; red: endothelial cells; purple: antigen-presenting cells, turquoise: B cells/plasma cells.
T lymphocytes
Ion channels are long known to be critically involved in the activation and effector function of immune cells. Excellent and detailed reviews of ion channels on T lymphocytes have been published elsewhere, which can be recommended for the interested reader [Cahalan and Chandy, 2009; Feske et al. 2012]. The best-studied candidates are probably the voltage-gated potassium channel KV1.3 (KCNA3) and the calcium-activated potassium channel KCa3.1 (KCNN4), the function of which has been found to be essential in triggering T-lymphocyte activation [Cahalan and Chandy, 2009]. Upon T-cell receptor (TCR) stimulation, an elevation of the cytoplasmic calcium concentration is mandatory for further T-cell activation [Lewis, 2001], and potassium channels maintain a negative membrane potential needed for longer lasting Ca2+ influx. Blockade of KV1.3 on T cells reduces effector functions, such as cytokine production and proliferation rates [Beeton et al. 2005, 2006]. In animal models of delayed-type hypersensitivity, type 1 diabetes, rheumatoid arthritis, and multiple sclerosis (MS), KV1.3 channel inhibitors ameliorated the respective disease courses [Beeton et al. 2001, 2011]. Initially, cells from KV1.3−/− mice did not show the expected phenotype due to compensating chloride currents [Koni et al. 2003], whereas later on, investigations showed a bias in KV1.3−/− mice towards an immunoregulatory phenotype leading to an ameliorated phenotype upon induction of experimental autoimmune encephalomyelitis (EAE) [Gocke et al. 2012]. High levels of KV1.3 expression were found on autoaggressive T cells isolated from patients with MS [Beeton et al. 2006]. The relative contributions of KV1.3 and KCa3.1 seem to differ on specific lymphocyte subsets and their activation levels. Naïve human T cells, effector memory T cells and probably also T helper 17 (TH17) cells depend on KV1.3, while the role of KCa3.1 seems to be more prominent on activated T cells, central memory T cells and TH1/2 cells [Feske et al. 2012]. Selective targeting of KV1.3 has attracted considerable attention as a potential novel treatment strategy for autoimmune diseases. Smaller companies collaborating with scientists active in this research field [Airmid (www.airmid.com) and Kineta Inc. (www.kinetabio.com)] as well as larger pharmaceutical companies (e.g. Amgen and Evotec) are performing preclinical studies with different KV1.3 blockers [Lam and Wulff, 2011]. However, none of these substances has so far advanced into clinical trials.
Inhibition of KCa3.1 using pharmacological blockers was effective in animal models of MS [myelin oligodendrocyte glycoprotein (MOG) peptide induced EAE] and rheumatoid arthritis (collagen antibody induced arthritis) [Chou et al. 2008; Reich et al. 2005]. Furthermore, KCa3.1 has been proposed to be a novel treatment for inflammatory bowel diseases (IBDs). KCa3.1−/− mice and mice treated with blockers showed an ameliorated diseases phenotype both in an adoptive transfer model and in a chemically induced IBD model [Di et al. 2010]. In contrast to KV1.3, targeting KCa3.1 for other indications has already been translated into clinical trials. However, the compound ICA-17043 failed in clinical trials of both sickle cell anaemia and exercise-induced asthma [Lam and Wulff, 2011]. Encouragingly, both studies showed that KCa3.1 blockade in humans is safe and well tolerated, opening up future possibilities for clinical trials of autoimmune and inflammatory disorders.
Apart from KV1.3 and KCa3.1, which possess either a voltage or calcium sensor, the recently discovered third group of potassium channels on T lymphocytes containing members of the two-pore domain (K2P) potassium channels is mostly time, voltage and calcium independent [Bittner et al. 2010b]. The K2P channels TASK1-3 (KCNK3, 5 and 9) are important for the activation and effector functions of T lymphocytes in vitro and in animal models of MS [Bittner et al. 2009; Meuth et al. 2008]. After induction of EAE with MOG peptide, TASK1−/− mice or mice treated with a novel TASK1 inhibitor showed a significantly ameliorated disease course, which was accompanied by a reduced activation of immune cells and less axonal damage compared with wild-type mice [Bittner et al. 2009, 2012]. Expression levels of TASK2 on T lymphocytes have been found to correlate with disease courses in patients with MS and rheumatoid arthritis [Bittner et al. 2010a, 2011]. In summary, a promising novel group of previously underestimated ion channels has entered the field of potassium channels on T lymphocytes. At the present time, the clinical benefit of targeting either one of the different potassium channels that control the membrane potential of these cells awaits further clarification.
As mentioned above, the level of intracellular calcium in T lymphocytes is tightly regulated due to the importance of intracellular calcium as central second messenger molecule. In the past couple of years, fundamental progress has been made towards identifying the molecular components of the Ca2+ signalling complex upon TCR stimulation. T lymphocytes mainly depend on store-operated calcium entry (SOCE) [Hogan et al. 2010]. TCR activation leads to the production of inositol-1,4,5-trisphosphate (IP3) and the release of Ca2+ from the endoplasmic reticulum Ca2+ stores. The ensuing activation of the calcium sensors stromal interaction molecule 1 (STIM1) and STIM2 results in an opening of calcium release-activated calcium (CRAC) channels. ORAI1 proteins appear to be the predominant isoform of CRAC channels in immune cells [Feske et al. 2012]. The importance of this pathway can be seen in human patients with a severe form of combined immunodeficiency, which occurs upon defects of CRAC channels and SOCE in T cells [Feske et al. 1996, 2006]. CRAC channels in T cells are crucial for autoimmune inflammation, which has, among others, been shown in animal models of MS and IBD in ORAI1−/−, STIM1−/− and STIM2−/− mice [Ma et al. 2010; McCarl et al. 2010; Schuhmann et al. 2010]. STIM1 and STIM2 are required for the proinflammatory function of both TH1 and TH17 cells in the context of EAE.
For further ion channels on T lymphocytes and thymocytes compromising ion channels with selectivity for different ions [e.g. purine receptor (P2X) channels, transient receptor potential melastin (TRMP) 2, TRMP7, Clswell, NaV1.5] we refer to recent excellent reviews by Cahalan and Chandy, and Feske and colleagues [Cahalan and Chandy, 2009; Feske et al. 2012]. In summary, pharmacological blockade of ion channels on T cells interferes with T-cell activation and effector functions and represents an attractive strategy for the treatment of T-cell-mediated autoimmune diseases. A clinical translation of this research concept, especially for T-cell potassium channels, is to be expected in the near future.
B lymphocytes
In contrast to ion channels on T lymphocytes, which have been long established as a potential target for the treatment of autoimmune diseases, the value of targeting ion channels on B lymphocytes is less clear. It is generally assumed that the main players on T lymphocytes also play a major role in B-cell physiology. The SOCE pathway involving STIM molecules and CRAC channels upon B-cell receptor activation seems to be comparable for B and T lymphocytes [Feske et al. 2012]. B cells from ORAI1−/−, STIM1−/− and STIM2−/− mice exhibit lower proliferation rates upon B-cell receptor activation [Gwack et al. 2008; Matsumoto et al. 2011]. While several B-cell functions seem to be barely affected by SOCE (e.g. B-cell development and antibody production), the failure of anti-inflammatory interleukin-10 production in B-cell specific STIM1−/−/STIM2−/− mice (Stim1f/fMb1cre/+, Stim2f/fMb1cre/+ and Stim1f/fStim2f/fMb1cre/+ mice) results in an exacerbation of EAE in vivo [Matsumoto et al. 2011]. Both KV1.3 and KCa3.1 are also expressed on B lymphocytes. Like T lymphocytes, B lymphocytes exhibit a switch in K+ channel expression from KCa3.1 to KV1.3 during the differentiation of naïve and early memory immunoglobulin D (IgD)+ B cells into class-switched memory B cells with high KCa3.1 and low KV1.3 expression [Wulff et al. 2004]. Respective channel blockade results in a reduction of B-cell proliferation. This implies potential therapeutic effects of KV1.3 and KCa3.1 blockers, while in vivo studies focusing on ion channel blockade on B cells in particular are still missing.
Interestingly, the discovery of K2P channel expression on T lymphocytes is paralleled by similar observations on B lymphocytes. TASK2 is expressed on mouse and human B lymphocytes [Bittner et al. 2010a; Nam et al. 2011] and is upregulated following B-cell receptor stimulation [Nam et al. 2011]. In addition, TREK2 (KCNK10) was identified as a second component of the K2P background current in mouse B cells [Zheng et al. 2009], while its contribution to B-cell function remains to be clarified. In summary, the therapeutic potential of specifically targeting ion channels on B lymphocytes cannot yet be evaluated.
Dendritic cells
Differentiation, antigen presentation and migration, which are key functions of dendritic cells (DCs), are influenced by a repertoire of different ion channels. DCs can be subdivided into heterogeneous and plastic subtypes involving resting immature DCs with high phagocytic activity in different organs (e.g. interstitial DCs or Langerhans cells) and mature DCs which present antigens and costimulatory molecules and migrate to draining lymph nodes [Banchereau et al. 2000]. All of the following descriptions apply only to conventional DCs of myeloid origin. Plasmacytoid DCs of lymphoid origin will not be taken into account, as no reports of ion channels on these cells exist so far.
The voltage-gated sodium channel NaV1.7 (SCN9A) is highly expressed on human immature DCs and especially CD1a+ DCs which differentiate into Langerhans cells on epithelial surfaces as shown by electrophysiological measurements [Kis-Toth et al. 2011; Zsiros et al. 2009]. Inhibition or siRNA-mediated knockdown of NaV1.7 results in a reduced cell migration underlining its importance for DC function. NaV1.7 expression is downregulated during DC maturation, and a concomitant upregulation of KV1.3 is observed. Apart from KV1.3, DCs express KV1.5 and KV1.3/KV1.5 heteromeric channels to different degrees [Mullen et al. 2006; Shumilina et al. 2007]. Comparable to T lymphocytes, these voltage-gated potassium channels regulate calcium homeostasis, and potassium channel blockers inhibit DC activation upon stimulation. The relative importance of these channels for DC maturation and activation is not completely understood, and DCs from KV1.3−/− mice were shown to be fully capable of activating wild-type T lymphocytes [Gocke et al. 2012]. Interestingly, KV1.3 and KV1.5 have been identified on blood-derived DCs that migrate into the central nervous system (CNS) and may exhibit anti-inflammatory properties in patients with MS [Mullen et al. 2006]. DC migration is regulated by chemokine signalling as binding of CCL19 and CCL21, for example, to the CCR7 receptor induced an intracellular calcium increase via both the IP3 pathway [Stolk et al. 2006] and ryanodine receptors [Uemura et al. 2007]. Different independent calcium channel systems have been described, not only compromising SOCE (including STIM2 and CRAC channel forming ORAI2 proteins) [Bandyopadhyay et al. 2011; Berridge, 2009; Hsu et al. 2001], but also L-type Ca2+ channels [Vukcevic et al. 2008]. This is in contrast to the SOCE pathway of lymphocytes which instead relies on STIM1 and ORAI1 [Shaw and Feske, 2012]. For an overview of calcium signalling in DC, see also Shumilina and colleagues [Shumilina et al. 2011]. Furthermore, the calcium-activated potassium channel KCa3.1 plays an important role in calcium-dependent cell functions, such as activation and migration, in a broad range of cell types including not only T cells, macrophages and mast cells but also DCs [Shao et al. 2011]. Apart from this, DC migration and other functions have been shown to require additional activity of other players, such as a calcium-activated chloride channel (ANO6) [Szteyn et al. 2012a], TRMP2 [Sumoza-Toledo et al. 2011], TRPM4 [Barbet et al. 2008], transient receptor potential V1 (vanilloid receptor, TRPV1) [Toth et al. 2009], aquaporins [Song et al. 2011; Wang et al. 2008], P2X7 receptors [Mutini et al. 1999], voltage-gated proton (Hv1) channels [Szteyn et al. 2012b] and acid-sensing ion channels (ASICs) [Tong et al. 2011].
Taken together, a complex and dynamic repertoire of ion channels orchestrates the different aspects of specific DC subtypes with varying functions. While some ion channels, such as KV1.3/KV1.5, KCa3.1 or SOCE, are considered well established, others have only recently been discovered. Further yet not described candidates can be expected in the future. Therefore, it is difficult to predict possible therapeutic exploitation strategies in the context of autoimmune neuroinflammation at the present time.
Astrocytes
Recent discoveries have drawn considerable attention to ion channels on astrocytes in autoimmune neuroinflammation. The inward-rectifying potassium channel KIR4.1 was proposed as a potential immune target in a subgroup of patients with MS [Srivastava et al. 2012] as about 50% of the patients in this cohort had elevated serum levels of KIR4.1-binding autoantibodies. Follow-up studies are currently underway to evaluate the diagnostic and therapeutic potential of these novel findings. Apart from this, reactive astrocytes within CNS lesions of patients with MS upregulate the voltage-gated sodium channel NaV1.5, different glutamate receptors and the P2X7 receptor [Black et al. 2010; Narcisse et al. 2005; Newcombe et al. 2008].
Neuromyelitis optica (NMO) is an autoimmune disease of the CNS that can be distinguished from MS by NMO-specific autoantibodies to aquaporin 4 (AQP4). AQP4 is a water channel which is densely expressed on astrocytic endfeet [Fujihara, 2011]. It was suggested that these antibodies are pathogenic and lead to destruction of the perivascular astrocytes [Parratt and Prineas, 2010], followed by lesion development and demyelination [Takano et al. 2010]. Preclinical studies are currently being performed aiming at a blockade of AQP4 antibody binding to astrocytes [Ratelade and Verkman, 2012]. Although no clinical trials for autoimmune neuroinflammation aiming specifically at astrocytic ion channels have been performed yet, considerable research attention concerning ion channel dysfunction on astrocytes can be expected in future.
Neurons
Several pathological ultrastructural changes are induced during the process of demyelination. Among them, myelin vacuolation, a process accompanied by degeneration of oligodendrocytes resulting in a splitting of the myelin sheath [Judge and Bever, 2006]. This loss of myelin is further enhanced by active stripping of the myelin sheath by macrophages and ‘dying-back gliopathy’ in which the inner tongue of the myelinating oligodendrocytes degenerates first [Ludwin, 2000]. As a consequence of these demyelination-associated mechanisms, paranodal and internodal voltage-sensitive potassium channels (KV channels, e.g. KV1.1) are exposed, downregulated (e.g. KV2.1), upregulated (e.g. KV1.4) or redistributed (e.g. KV1.2/KVβ2) [Jukkola et al. 2012], leading to abnormal potassium outward currents associated with slow action potential conduction, conduction failure or changes in the axon’s capacity to repetitively discharge [Dunn and Blight, 2011] due to membrane hyperpolarization towards the potassium equilibrium potential (approximately 100 mV, see Figure 2). In addition, demyelination leads to loss of structural proteins anchoring KV channels in the membrane (e.g. contactin and Caspr2) [Gu and Gu, 2011; Horresh et al. 2008; Ivanovic et al. 2012] accompanied by a drift of KV channels to other sites of the axon, inducing a reduction in resistance, slowing of conduction velocity and conduction failure [Boyle et al. 2001]. Pharmacological inhibition of exposed KV channels at demyelinating axons was hypothesized as an attractive strategy to improve action potential generation in demyelinating disorders [Sedehizadeh et al. 2012]. Consequently 4-aminopyridine (4-AP), a well-known potassium channel blocker, was improved by providing a sustained release formulation and tested for mobility improvement in patients with MS versus placebo. Interestingly, 4-AP-mediated improvement in conduction translates into clinical benefits, as shown by objectively (e.g. on the 25 ft walk test) and subjectively (e.g. rating scales) measuring mobility in patients with MS compared with placebo in a phase III trial [Goodman et al. 2009]. A further multicenter, double-blind phase III trial included patients with definite MS of any disease course, and participants were randomized to 9 weeks of dalfampridine (10 mg twice daily) or placebo. The primary outcome of the study was defined as percentage of timed walk responders (TWRs; defined as consistent improvement on the timed 25 ft walk) in each treatment group. The percentage of TWRs was significantly higher in the dalfampridine group (42.9%) compared with the placebo group (9.3%, p < 0.0001) [Goodman et al. 2010]. Based on this class 1 evidence, dalfampridine extended-release tablets were approved for patients with MS, of any disease course, with impaired mobility. However, beneficial clinical effects were explained by an improvement in action potential conduction by inhibition of KV channels on demyelinated axons, though average plasma concentrations of 0.243 ± 113 μM reached in human subjects (therapeutic dose of 10 mg twice daily) [Goodman et al. 2007] are three to four orders of magnitude lower than IC50 values (concentration at which 50% of the current is blocked) of these channels (e.g. KV1.1) [Judge and Bever, 2006]. Of note, mechanisms of KV current inhibition by 4-AP depend on factors such as gating processes of channel activation, deactivation and inactivation, the frequency of stimulation and the kinetic state of the channel [Choquet and Korn, 1992; Kirsch and Drewe, 1993; McCormack et al. 1994; Yeh et al. 1976]. Due to the redistributed channel expression along demyelinating axons and the modified expression of channels and subunits, sensitivity to 4-AP may increase [Fehlings and Nashmi, 1996; Karimi-Abdolrezaee et al. 2004; Shi et al. 1997].

(a) Changes in potassium channel expression and distribution in multiple sclerosis. Demyelination induces increased exposure of fast K+ channels (KV1.1), which leads to membrane hyperpolarization, an increased threshold for successful action potential transmission, and an increased likelihood of conduction failure. In addition, demyelination causes a loss of structural proteins (e.g. contactin, Caspr2) anchoring K+ channels in the membrane. In consequence, channels drift to other places of the axon, thereby lowering the resistance of the axon and conduction velocity, resulting finally in conduction failure. (b) ASIC1 channels contribute to axonal degeneration. During inflammation activation of T cells, microglia cells and macrophages can cause production of nitric oxide (NO) and free oxygen radicals, leading to mitochondrial damage with subsequent energy failure. As a consequence, emerging tissue acidosis leads to activation of ASIC1 with intracellular accumulation of Na+ and Ca2+ ions. Increased [Ca2+]i activates secondary injury cascades (e.g. proteases, break down of the cytoskeleton), resulting in axonal degeneration.
Increased influx of extracellular calcium in neurons or axons via voltage-gated calcium channels (VGCCs) was assumed to contribute to axonal/neuronal degeneration associated with neurological impairment. The pore-forming α1B subunit (CaV2.2, N-type calcium channels) was found to be overexpressed in active MS and EAE lesions [Kornek et al. 2001]. In addition, two L-type (CaV1.2, CaV1.3, CaV1.4) calcium channel inhibitors (bepridil, nitrendipine) showed beneficial effects in adoptive transfer experimental autoimmune encephalomyelitis (AT-EAE) mice by reducing inflammation and axonal pathology [Ehling et al. 2011]. These results are of interest but there are currently no planned or ongoing clinical trials using VGCC inhibitors most likely due to the expected side effects, for example, cardiac side effects.
Very recently, Schattling and colleagues were able to identify that TRPM4 nonselective cation channel contributes to axonal and neuronal degeneration in EAE and MS [Schattling et al. 2012]. Functional TRPM4 expression could not only be detected in murine and human neuronal somata, but also in axons in autoimmune inflammatory CNS lesions in mice and in active human MS lesions. Using Trpm4−/− mice or pharmacological inhibition of the channel (e.g. with glibenclamide) resulted in a significantly ameliorated disease course accompanied by reduced axonal and neuronal degeneration, although disease initiation and peak disease were indistinguishable from control animals. As demonstrated in vitro and in bone-marrow chimera experiments, these beneficial effects were independent of EAE-relevant immune functions, opening up a chance for a new directly neuroprotective therapeutic strategy (see Figure 2). A functional link between ASIC1 and TRPM4 channels is given because, under inflammatory conditions, ASIC1 channels mediate Ca2+ influx in the axon in a pH-dependent manner, which, in turn, activates TRPM4, thereby allowing a sodium influx into the axon with activation of secondary injury cascades.
An emerging body of evidence is available, suggesting that sodium channels may represent promising therapeutic targets in autoimmune-mediated disorders (reviewed by Eijkelkamp and colleagues and Waxman [Eijkelkamp et al. 2012; Waxman, 2008]). Early evidence has shown that axonal degeneration involves persistent sodium influx through noninactivating sodium channels [Craner et al. 2003]. When this influx overwhelms the ability of the adenosine triphosphate (ATP)-dependent Na+/K+-ATPase pump to shuffle sodium ions to the extracellular space, the Na+/Ca2+ exchanger operates in a ‘reverse mode’, thereby importing calcium ions into the axon (for the detailed mechanism, see Figure 2). In turn, elevated intra-axonal Ca2+ concentrations further increase currents through NaV1.6 channels, thereby providing a positive feedback loop, which results in significantly increased Ca2+ concentrations. Activation of cell death cascades is a result of the heightened Ca2+ content. Of note, this injurious cascade can occur in myelinated axons with clusters of voltage-gated sodium channels at the nodes of Ranvier due to energy failure, but they are especially prominent in MS lesions, where demyelinated axons express an upregulated number of sodium channels (Figure 2) [Waxman, 2008]. Using sodium channel inhibitors (e.g. tetrodotoxin, lidocaine, phenytoin or carbamazepine) at concentrations that do not compromise action potential conduction, axon protection against anoxic and ischemic conditions could be achieved in vitro [Fern et al. 1993; Stys et al. 1992]. These findings provided a rationale to examine whether application of sodium channel blockers, such as phenytoin or carbamazepine, in EAE (see Table 1) could preserve the axon’s functionality and integrity. These blockers indeed enhanced action potential conduction along demyelinated axons, which was associated with reduced immune infiltrates in the animal’s CNS. However, withdrawal of phenytoin or carbamazepine resulted in acute disease exacerbation of EAE mice (see Table 1), significant lymphocyte infiltration into the CNS and mortality [Black et al. 2007; Kieseier and Hartung, 2013]. These safety concerns dampened the enthusiasm to further clinical development of sodium channel blockers for MS, despite the fact that a randomized, double-blind placebo-controlled parallel-group trial in 120 patients with secondary progressive MS was conducted using lamotrigine (400 mg/day) over 24 months [Kapoor et al. 2010]. The effect of lamotrigine on cerebral volume of patients with secondary progressive MS did not differ from that of placebo over 24 months. Very recently, another member of the voltage-gated sodium channel family could be linked to cerebellar dysfunction, namely NaV1.8. Ectopic expression of this channel in the cerebellum resulted in altered electrophysiological properties of Purkinje cells, disrupting coordinated motor behaviour. Use of a NaV1.8-specific inhibitor demonstrated that EAE symptoms can partially be reversed [Shields et al. 2012].
Summary of clinical efforts targeting neuronal ion channels.
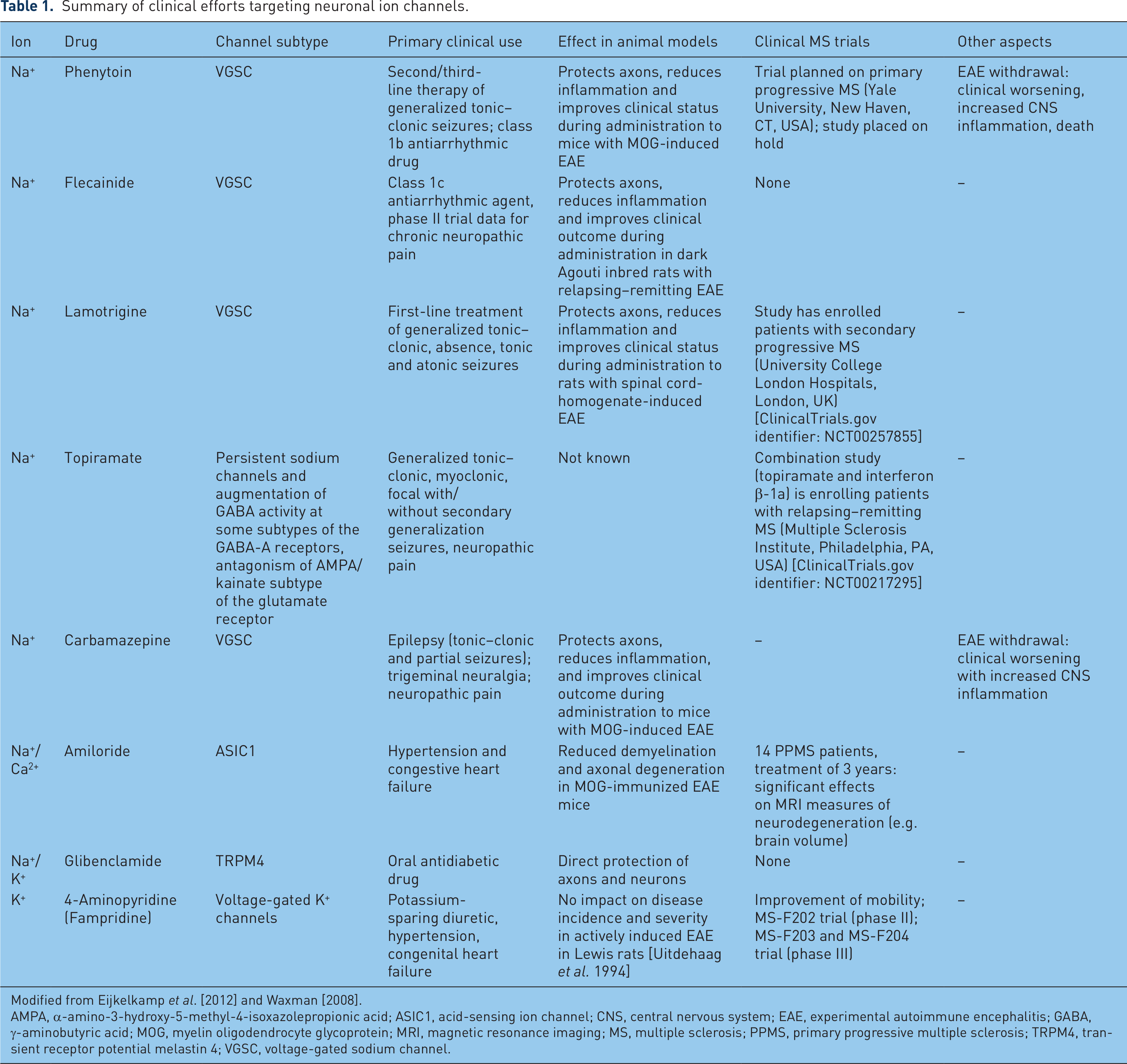
Modified from Eijkelkamp et al. [2012] and Waxman [2008].
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; ASIC1, acid-sensing ion channel; CNS, central nervous system; EAE, experimental autoimmune encephalitis; GABA, γ-aminobutyric acid; MOG, myelin oligodendrocyte glycoprotein; MRI, magnetic resonance imaging; MS, multiple sclerosis; PPMS, primary progressive multiple sclerosis; TRPM4, transient receptor potential melastin 4; VGSC, voltage-gated sodium channel.
Voltage-gated sodium channel activity has also been shown to determine T-cell development [Lo et al. 2012], T-cell activation [Lai et al. 2000] and their invasiveness [Fraser et al. 2004], also suggesting an anti-inflammatory potential for sodium channel modulation.
The family of ASICs consists of several isoforms, but in the CNS, the predominant functional channel subunit is ASIC1a, which mediates Na+ and Ca2+ influx, thereby contributing to axonal degeneration [Wemmie et al. 2002; Xiong et al. 2008]. Direct evidence implicating ASIC1a in neuronal injury has been shown in experimental stroke [Vergo et al. 2011; Xiong et al. 2004], 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine induced Parkinson’s disease [Arias et al. 2008] and in an animal model of MS [Friese et al. 2007]. In the EAE model, Asic1−/− mice showed significantly reduced clinical deficits and axonal degeneration compared with control animals after MOG immunization. Amelioration of axonal degeneration was observed in the model, although markers of inflammation were comparable between both groups, pointing towards a direct impact of ASIC1 on axonal injury rather than an indirect one mediated by anti-inflammatory mechanisms. Of note, ASICs are expressed on immune cells. In addition, Friese and coworkers were able to show beneficial effects in the EAE model by using the potassium-sparing diuretic amiloride as an ASIC1 inhibitor [Friese et al. 2007]. These results could also be shown in relapsing–remitting animal models of MS when amiloride was applied after the first relapse [Vergo et al. 2011]. Axonal expression of ASIC1 in human spinal cord and optic nerve tissue samples provided further data to challenge the concept of ASIC1 inhibition in a translational study. Arun and colleagues reported the result of an open-label 3-year trial to evaluate neuroprotective effects of amiloride in patients with primary progressive MS (PPMS) [Arun et al. 2013]. First, these confirmed an increased expression of ASIC1s in axons within chronic inactive postmortem lesions from patients with PPMS. Second, they tested the neuroprotective effect of amiloride in a cohort of 14 patients with relapsing–remitting MS using magnetic resonance imaging (MRI) markers of neurodegeneration as outcome measures (whole-brain volume and tissue integrity as assessed by T1-weighted and diffusion tensor MRI). Patients underwent serial MRI scans before (pretreatment phase) and during (treatment phase) amiloride treatment for a period of 3 years. The authors observed a significant reduction in the normalized annual rate of whole-brain volume and in diffusion indices of tissue damage within the white and deep grey matter structures during the treatment phase compared with the pretreatment phase. They claim this represents another piece of evidence for the contribution of ASIC1 to neurodegeneration in MS and suggest that amiloride may exert neuroprotective effects in patients with PPMS. Amiloride was given orally at a daily dosage of 10 mg which is known to have a diuretic effect by blocking renal epithelial Na+ channels in the distal tubule, resulting in excretion of water, sodium and chloride, and retention of potassium, with the danger of metabolic acidosis. Blood tests for urea and electrolytes were performed (but not reported) and there was no control for systemic (and subsequent cerebral) changes of acid-base metabolism and volume. Hence, an unspecific effect of amiloride on the MRI parameters used to evaluate neuroprotection cannot be excluded, and further studies taking these caveats into account are warranted.
Outlook
In summary, pharmacological targeting of ion channels is being increasingly recognized as an attractive and feasible strategy for the treatment of autoimmune diseases. But broad expression pattern of most ion channels and their partial functional redundancy together with a lack of highly specific pharmacological inhibitors makes it difficult to translate concepts of specific ion channel subtype targeting from bench to bedside. Combined efforts of basic researchers, pharmaceutical companies and clinicians will be necessary to achieve this goal in the future.
Sodium channels were identified as interesting candidates for clinical translation but results from withdrawal experiments in mice and the negative lamotrigine trial in patients with secondary progressive MS somehow lower the level of enthusiasm. Potassium channels are interesting candidates and KV1.3 inhibitors are under preclinical investigation by different companies. However, the specific KCa3.1 blocking compound ICA-17043 failed to show efficacy in clinical trials of sickle cell anaemia and exercise-induced asthma. So far no trial was designed to test KV1.3 or KCa3.1 inhibitors in autoimmune inflammatory disorders. Two pore domain potassium channels were recently described as potential target structures for the treatment of T-cell mediated neurodegeneration and specific inhibitors are currently under development, but this concept is still in the preclinical development phase.
ASIC1 and TRPM4 channels represent two new interesting candidates, which are functionally linked. ASIC1s mediate Ca2+ influx in the axon in a pH-dependent manner, which, in turn, activates TRPM4, thereby allowing a sodium influx into the axon with activation of secondary injury cascades. First results from a smaller cohort study using the ASIC1 inhibitor amiloride demonstrate a neuroprotective effect in 14 patients with MS using MRI markers of neurodegeneration as outcome measures (whole-brain volume and tissue integrity as assessed by T1-weighted and diffusion tensor MRI).
Footnotes
Acknowledgements
We thank Heike Blum for excellent work on graphical design and illustration of the figures.
Funding
This work was supported by the Else-Kröner-Fresenius Stiftung (SB and SGM).
Conflict of interest statement
The authors have no conflicting financial interests.